Radioactive isotopes for research, medicine, technology
1.
Nuclear and radiation physics
1.0. Physics - fundamental natural
science
1.1. Atoms and atomic nuclei
1.2. Radioactivity
1.3. Nuclear reactions and nuclear
energy
1.4. Radionuclides
1.5. Elementary particle and
accelerators
1.6. Ionizing radiation
1.4.
Radionuclides
In this chapter we will approach the origin and properties
of the most important radionuclides - important
either in terms of physical or general science, or radionuclides
used in various fields of science and technology, including
medicine, in industrial applications, radionuclides occurring in
the environment, etc. Mendeleev's periodic table contains 118 now
known elements. However, there are significantly more
radionuclides than stable nuclides, a total of over 2000 are
known. Within a single element, several radioisotopes
are usually represented, sometimes even ten or more.
Natural radionuclides
In addition to stable chemical elements, radioactive
elements of natural origin also occur in very low
concentrations in the natural environment. These natural
radionuclides can be divided into three categories according to
their origin and formation :
Primary (primordial) radionuclides ([great]original, fossil)
These are radionuclides that formed together
with other (stable) nuclei of heavier elements during
cosmic nucleogenesis by thermonuclear reactions
in the interior of stars
(as mentioned in §1.1. in the paragraph "Cosmic
alchemy" - "We are the descendants
of the stars!"), which then exploded as supernovae and enriched
with these elements the germinal cloud from which our Sun
and the solar system formed. They became part of the
Earth during the formation of the solar system about 4-5
billion years ago, the Earth to bring them from
space. To date, however, only those
radionuclides have been preserved that have a very long
half-life - greater than about 108 years.
Note:
Some primary radionuclides have, among other things, geological
significance. The heat released by the
radioactive decay of natural radionuclides,
uranium 235,238U, thorium 232Th and potassium 40K, is probably an important source of geothermal
energy, heating the Earth's interior (see below "Geological significance of natural
radioactivity").
The most common primary
radionuclide in terrestrial nature is potassium 40K, whose average content in the
earth's crust is about 3.10-3 %. Potassium 40 decays with a half-life of T1/2 = 1.27.109 years to beta--
radioactivity to argon 40Ar (89%) and electron capture to calcium 40Ca (11%) - see below " Potassium - 40-K "; both of
these daughter isotopes are stable, further decay no
longer continues.
Another widely used natural primary
radionuclide is thorium 232Th (concentration
in the earth's crust approx. 8-12.10-6), which with a half-life
T1/2 = 1.39.1010 years decays
by alpha-radioactivity (in
combination with beta) gradually
into a series of radionuclides of the so-called thorium
series (Fig.1.4.1 left).
However, the most important
natural radionuclides of this primordial origin in the
earth's crust are uranium 238U (average concentration in the
earth's crust approx. 2-4.10-6, T1/2 = 4.51.109 years), and uranium 235U (concentration approx. 2-3.10-8, T1/2 = 7.1.108 years). Both
of these isotopes of uranium with alpha-decay (in combination with beta) are
gradually converted into a series of radionuclides of
both uranium decay series - Fig.1.4.1 in the middle and
right, is described below.
Note: The element uranium,
first isolated in pure form in 1841, got its name from
the then newly discovered planet Uranus (which
in turn was named after the god Uranus in ancient Greek
mythology). It is a heavy silvery metal (density 19 g/cm3), oxidizing
in air with a dark gray coating. It was used to color
glass. In 1896, radioactivity was first discovered in
natural uranium ore by H. Beckerel. Due to its high
density, 238U ("depleted uranium") is also used
for some "non-nuclear" applications (eg
projectiles with high penetration, or for shielding).
However, uranium has the main use in nuclear
technology as fissile material -
see §1.3 "Nuclear reactions", especially the
section "Fission of atomic
nuclei".
Other
primordial radionuclides with long half-lives also occur
in small trace amounts in nature: chlorine 36Cl (T1/2= 3.105 years),
beryllium 10Be (T1/2 = 1,52.106 years), lutetium 176Lu (T1/2 = 3,8.1010 years), rhenium 187Re (T1/2 = 4,16.1010 years), rubidium 87Rb (T1/2 = 4,88.10 10 years), samarium 147Sm (T1/2 = 1,06.1011 years), .... Some of them are used for
radiometric dating of rocks (see
"Radioisotope dating" below).
Note:
Insignificant from the
radiation point of view are some isotopes with an
extremely long half-life (usually between 1014 and 1018 years),
whose radioactivity is often not even safely proven - eg 48Ca, 64,70Zn, 76Ge, 82Se, 92,100Mo, 116Cd, 124Sn, 123Sb, 136,142Ce, 144,145,150Nd, 147-149Sm, 152Gd, 156Dy, 165Ho, 180,182,183,186W,
192Os,
190,192,198Pt, 196Hg, 209Bi.
 |
Fig.1.4.1. Natural decay
series of thorium 232Th and uranium 235U and 238U .
The Neptunium decay series is shown
below in the passage "Actinium 225 Ac", in the diagram at the top left.
Sorry: ... the picture is
unfinished! -they will come to add halves, energy
+ graphic design ...... |
Secondary radionuclides
- decay products
of primary radionuclides
By the decay of primary heavy radionuclides, a whole
series of secondary radionuclides is
created continuously. The natural radionuclides 232Th, 238U and 235U are
transformed by alpha (and later even by beta) decay into
nuclei, which are also radioactive, as well as their
further and further transformation products. We say that
these radionuclides generate radioactive decay
series - chain (decay chain), where the
individual daughter products show both alpha and beta
radioactivity and the excited nuclei emit gamma
radiation. The so-called secular radioactive
balance is established in these decay
series (see §1.2
"Radioactivity").
In the terminology of
the passage "Stability and instability
of nuclei" §1.2
"Radioactivity", it can be said that heavy
thorium and uranium nuclides "jump" down
the energy levels and "rolls" into the valley
of stability, until they reach an equilibrium
configuration of lead or bismuth.
Radioactive
decay series
There are three radioactive decay series in nature
(Fig.1.4.1): decay series of thorium232Th (left), decay series of uranium 238U (uranium-radiun series - it includes the radium 226Ra - in Fig. right) and
decay series of uranium 235U
(middle) called uranium-actinium
series *). The neptunium 237Np decay series can be
artificially obtained (shown below
in the passage "Actinium 225 Ac",
the picture top left).
*) Actinium 227Ac is a part
of the decay series of uranium-235. In the older
literature, 235U it was often called actinouran and
its decay actinium series.
These decay chains of heavy nuclei
are somewhat similar :
¨ Primary parent nuclides have very long
half-lives - millions to billions of years. As a
result, a permanent (long-term) so-called secular
radioactive equilibrium *) is established in the
decay series (see §1.2, section
"General laws of atomic
nucleus transformation").
*) More precisely, this would be the case with
hermetically sealed preparations. In
nature, this one is the equilibrium
is disturbed by the escape of some isotopes of
the decay series (especially radon gas) from the deposit.
¨ The decay series are isotopes
of heavy elements showing mostly alpha
radioactivity (to a lesser extent also beta).
¨ The nucleon (mass) numbers of
the members of the series correspond to the formulas (n
is an integer): 4.n for the thorium
series, 4.n+1 for the neptunium series, 4.n+2
for the uranium series 238U and 4.n+3 for 235U series
(actinium). This is due to the fact that a-decay
reduces the nucleon number by 4 ( b -decay does not
change them). It follows from this regularity that there
are exactly 4 basic independent decay
series.
¨ About halfway trought the series, the isotopes
of gaseous radon (formerly called emanation)
occurs.
¨ Some daughter nuclide (such as 211, 212, 213Bi
and 227Ac) are converted by both to a and b radioactivity,
leading to branching of the decay chain.
The decay b is then followed by the decay a and the
decay a vice versa the decay b, so that the two
branches merge again.
¨ Natural decay series end with stable isotopes of
lead, neptunium series with bismuth.
Natural decay series are
elaborated in more detail in Fig.1.4.1, in summary they
can be written as follows :
232Th
® 6 a + 4 b + 208Pb ; 238U ® 8 a + 6 b + 206Pb ; 235U ® 7 a + 4 b + 207Pb ; 237Np ® 7 a + 4 b + 209Bi .
The Neptunium decay series is drawn in more detail below
in the figure in passage Aktinium-225.
The details of the transformations
of individual radionuclides in decay series are quite
complex and do not fit into draw them in Fig.1.4.1. Some
nuclides of decay series are pure emitters a (eg 232Th, 238U, 216Po, ......)
or b (eg 228Ra, 206Tl, ........), however, most nuclei are
transformed into excited levels of daughter nuclei and
therefore emit also g
radiation (they are eg 228Ac, 214Pb, 214,212Bi, 210Tl, .....
.and others, but for some g is weakly
represented, eg for 222Rn, 239Pu, ...).
Decay series of transuranics
Analogous decay series are also for
artificially produced transurans, which
ultimately follow to the mentioned basic series. E.g. 241Am ® a + 237Np ® 7 a + 4 b + 209Bi - neptunium
decay series; 239Pu ® a + 235U ® 7 a + 4 b + 207Pb - 235U-actinium decay series; 252Cf ® a + 248Cm ® a + 244Pu ® a + 240U ® b + 240Np ® b + 240Pu ® a + 236U ® a + 232Th ® 6 a + 4 b + 208Pb - thorium
decay series. In the case of heavy transuraniums, this is
accompanied by the spontaneous fission of
nuclei (§1.2, section " "Exotic "types of
radioactivity").
Cosmogenic radionuclides
These are natural radionuclides that are continuously
formed by nuclear reactions during the passage
of high-energy cosmic radiation (its
secondary components) through the Earth's atmosphere.
This includes especially radiocarbon 14C and tritium 3H, in very small quantities
arise some other cosmogenic radionuclides - e.g. 7,10Be, 32P, 35S, 36Cl. The
mechanism of formation of cosmogenic radionuclides is
outlined in §1.6, part "Cosmic radiation", Fig.1.6.7.
Cosmogenic radiocarbon 14C is used in
archeology for radiometric age determination of
objects of biological origin (see "Radioisotope dating"
below).
To sum up, long- lived
radioisotopes with a half-life of more than about 100
million years occur in our terrestrial nature. Of the shorter
ones, only those that are continuously created by natural
processes - cosmic radiation or in the decay chains of long-term
isotopes. All other short-lived radioisotopes, formed during
stellar nucleogenesis in large quantities (especially
in supernovae), have not been preserved on
Earth - they are sometimes called extinct, defunct,
vanihed radionuclides; their former existence is
evidenced by the observed surplus of stable products of their
decay (cf. below "Radiometric dating", passage "Dating using decayed
radionuclides"). Such
important extinct radioisotopes are iodine 129 I, aluminum 26Al, and iron 60Fe, whose
radioactivity may have been involved in the formation of the
protoplanetary disk, planets, and asteroids at the beginning of
the solar system's development.
Radioactivity
in the natural environment
All nature (terrestrial and space) contains a small
amount of radioactivity, especially the above-mentioned
natural radioactive elements. Compounds of primary and secondary
radionuclides (radioactive decay series) enter the soil,
water and air (especially through gaseous radon) from
rocks during their erosion. During the combustion
of coal, some natural radionuclides are released into the air (especially radon 222Rn, polonium 210Po and lead 210Pb), which accumulate at greater
depths due to the decay of the primary natural radionuclides
uranium and thorium. During volcanic activity,
the ejected clouds of volcanic smoke and dust contain a
significant amount of radon radionuclides, polonium-210, lead-210
from great underground depths. Compounds of cosmogenic
radionuclides also enter the biosphere from the atmosphere. From
soil, water and air, natural radionuclides get into plants and in
the food chain into animals and humans.
In our environment, therefore, we are
constantly exposed to ionizing radiation from
cosmic rays and natural radionuclides in the air, in surrounding
objects, in soil and rocks, building materials, even in our
bodies.

Fig.1.4.2. Scintillation spectrum of gamma radiation of clay (sample of 200g of arable land from Central Moravia). Weak gamma peaks from the secondary radionuclides of
the uranium and thorium decay series (isotopes of bismuth,
thallium, lead) and a relatively significant 1460 kV peak of the
primary radionuclide potassium 40-K are evident.
In recent decades, this has been
approached by radiation exposure to artificial
radiation sources - X-rays, accelerators, artificial
radionuclides. Especially in targeted use in medical
radiodiagnostics and radiotherapy, and possibly also from
unwanted radioactive contamination (radiation
accidents, nuclear waste) - §5.2, part "Sources of radiation by ionizing
radiation". The favorable
and unfavorable biological effects of that natural and artificial
radiation are discussed in detail in §5.2 "Biological effects of ionizing radiation".
Geological
significance of natural radioactivity
Although the relative content of natural radionuclides in the
"building" material of the Earth is not high, their
total amount in the Earth's interior and crust is so large, that
the energy released by them can have
considerable geothermal significance.
According to current geological
knowledge, our planet Earth consists of several
layers. The solid surface consists of a relatively thin layer of earth's
crust, the so-called lithosphere (Greek lithos = stone),
about 5-50 km thick (depending on whether we are in the sea,
land, mountains). The Earth's crust is not solid, but is
"broken" into individual lithospheric plates which
move slowly relative to each other. Below it lies the Earth's
mantle (further divided into the upper and lower
mantle), which is to some extent malleable (plastic) and forms
the largest part of the volume and weight of the globe. It
consists of partially molten rocks with a temperature of about
600-1250 °C. Thermal convection takes place in the
mantle, through which heat is transferred from the inner part of
the Earth to the surface. The plasticity of the mantle, on which
the individual parts (plates, "floes") of the earth 's
crust "float", and the convection of heat is the
driving force of continental shifts, plate tectonics and volcanic
activity *). Inside the Earth is the Earth's core
(Moon size) in which the temperature is likely to exceed 4000
°C; its outer part is fluid, the inner core at the very center
of the Earth is (according to indirect geological measurements)
solid. The solid iron inner core of the Earth "floats"
and rotates in the liquid outer core, which generates a
geomagnetic field. This magnetic field creates a
"shield" against ionizing cosmic radiation and the
solar "wind". Without him, life on the planet would not
be possible.
*) These are "ordinary" volcanoes
located in places where lithospheric plates collide. However,
beneath the thin shell of the earth's crust, in the earth's
mantle, like a "time bomb", massive streams of
hot molten rocks rise by thermal convection . If it
melts to the surface, under great pressure they erupt as
destructive supervolcanoes .
Total heat flux from the Earth's interior is
estimated at about 45 Terawatts *). Such a massive heat flux is
difficult to explain by the heat remaining inside the Earth (due to the long time of about 4.5 billion years since
the Earth's origin), or tidal gravitational
action during the Earth's rotation, the Moon's orbit around the
Earth and the Earth around the Sun (§1.2,
passage "Gravitational gradients -
tidal forces" in the book
"Gravity, black holes ..."). The radioactive
decay of natural radionuclides could contribute about
60% to the development of heat in the Earth's interior. After
8-10 TW, the decay series of 232Th and 238 + 235U isotopes could supply, 40K of radioactive potassium would contribute 3-5 TW of
heat flux. Without heat from naturally occurring radioactivity,
the Earth's core would have cooled and solidified billions of
years ago, which would probably have prevented the origin and
development of life on Earth ("Anthropic
Principle or Cosmic God", part
"Stars, Planets, Life") ..!..
*) The average density of geothermal heat
flux at the Earth's surface is about 65-100 mW/m2, which is less than
ten thousandths of the intensity of incident solar radiation.
It has also been hypothesized that a natural
fission nuclear reactor similar to the one found in the Oklo
mine could operate inside the Earth (see
§1.3, section "Nuclear reactors", section "Natural nuclear reactors?
"). Billions of years ago, when the
concentration of fissile uranium 235U was still high enough, this is quite likely. In order
for such a reactor to function even at the present time, when the
share of fissile 235U considerably decreased, the concentration of uranium
in the material inside the Earth would have to be significantly
higher than in the rocks we know of the earth's crust; about it
we have no evidences.
We have relatively little information
obtained indirectly about the structure of areas inaccessible to
us. Important data on the distribution of radioactive elements in
the Earth's interior could be obtained by detecting
neutrinos, so-called geoneutrinos, formed
during b- radioactive decay of natural radionuclides, which freely
pass from the Earth's interior through thick rock layers to the
surface (see §1.2, section "Neutrinos - "ghosts"
between particles",
passage "Neutrinos from natural radioactivity").
Radioisotope
(radiometric) dating
The independence of the rate (half-life) of radioactive decay
from external conditions *) allows the use of the exponential decay
law of radioactivity as a kind of clock - a natural
"chronometer".
*) Situations where the course and rate of
radioactive decay may depend somewhat on external radiation or
chemical factors are discussed in §1.2, section "Independence of radioactive decay
on external conditions".
The basic idea of using radioactive decay
to measure time is simple. If we know the initial number N0 (at time t = 0) of
radioactive nuclei of the parent nuclide X with a known
half-life T1/2 in the sample, then by measuring the absolute activity
A(t) of the sample, or the ratio of the concentration of parent X
and daughter Y isotopes, at some later time t we
can determine from the decay law this time t , elapsed
from the sample (t = 0) to the time of measurement - thus the age
of the sample. From the decay law of radioactivity (derived in §1.2, part "General laws of transformation of
atomic nuclei") NX(t)
= NX(0).e-l.t
= NX(0).e-t.(ln2/T1/2) = NX(0).2-t
/T1/2
; NY(t) =
NX(0).[1-e-l.t]
= NX(0).[1-e-t.(ln2/T1/2)] = NX(0).[1-2-t
/T1/2],
for the sought age t the formula follows
t = T1/2 . [1
+ NX(t) /
NY(t)] ,
where NX(t)
is the current amount of the parent element X and NY(t) is the current
amount of the daughter element Y in the test sample (the initial amount of NX(0) of the parent radionuclide is usually unknown,
sometimes we try to estimate it, eg at 14C).
All materials (on Earth and in space)
contain a small amount of natural radionuclides
with long half-lives. The rates of their radioactive
transformation can, under certain conditions, be used to determine
the ages of various objects and samples of materials.
This method of determining the age of samples is generally called
radioactive, radioisotope or radiometric
dating, or radioisotope chronometry;
according to the investigated nuclides then eg radiocarbon
dating (using carbon-14, described
below), potassium-argon or uranium-lead
dating. It is used in archeology, paleontology, geology (geochronology)
and astronomy (age of meteorites and planets).
There are two basic conditions that should
be met for accurate radioisotope dating :
1. The initial conditions are known -
the concentrations or ratios of the parent and daughter isotopes
at the time of the natural origin of the investigated material
(at time "0");
2. The investigated materials must be
closed or isolated systems from their inception,
so that no parent or daughter isotopes have escaped or entered
them.
These conditions are never 100% met, so
"straightforward" radiometric dating is not completely
accurate. It will be outlined below which methods can be used to
perform independent calibrations and increase the
accuracy and reliability of radioisotope dating (so-called isochronous and cocondon
dating is described).
Radiocarbon dating method (also called carbon chronometry)
using radiocarbon 14C, which is formed by the action of
neutrons ejected by cosmic radiation from the nuclei of atoms, on
nitrogen in the upper layers of the Earth's atmosphere: no + 14N7 ® 14C6
+ p+ (see §1.6, section "Cosmic radiation", Fig.1.6.7). This creates
about 2 atoms of 14C per second per 1 cm3 of atmosphere. Carbon 14C, as a long-term radionuclide (T1/2 = 5730 years, pure b-, energy 158keV) constantly contaminates
the biosphere, oxidizes to 14CO2 in the atmosphere, enters the biocycle - the food chain
of living organisms on Earth. Through photosynthesis, it gets
from the atmosphere into plants, from there through food into the
bodies of animals. It is therefore contained in all living
organisms. The concentration of 1 atom of 14 C is stabilized to about 8.1013 atoms of ordinary 12C; one gram of natural carbon in all living organisms
contains an activity of about 0.25 Bq 14C. After the organism dies, its metabolic contact with
the atmosphere and the supply of 14C are interrupted, so that the concentration of radium
carbon begins to decrease by its radioactive b -decay 14C ® 14N + e- + n´ with a half-life of 5730 years. This
also changes the relative proportion between the carbon isotopes 14C, 13C and 12C.
From the ratio between the relative
proportion of radioactive isotope 14C and stable isotopes of carbon in the studied
historical object of biological origin (eg wood,
remains of organisms, etc.) we can approximately determine the age
of this object - the time that has elapsed since the death of the
organisms from which the object originated. This
method was first used by W.F.Libby in 1946.
In the simplest case, we measure the specific
activity of 14C in the investigated historical object of biological
origin and compare it with the equilibrium specific activity of
radiocarbon in the atmosphere and in living organisms. Sensitivity
of 14C
activity measurements using proportional or
scintillation detectors with liquid scintillators (see §2.6,
section "Liquid scintillators"), due
to the half - life of 14C 5730 years, limits the time range of this method to
about 30 000 years. Substantially lower values of 14C/12C can be determined
by mass spectrometry (see §2.6 , section "Magnetic spectrometers").
The precondition for the correctness of radiocarbon chronometry
is that the ratio of 14C/C12 in the past, was always the same during the period from
which the measured samples originated. In order for this ratio to
remain constant, the intensity and composition of cosmic rays and
their effects on the upper atmosphere must remain constant. The
primary cosmic rays coming from interstellar space are likely to
be constant for at least 105 years. However, the component coming from the Sun is
time-varying depending on solar activity (occurrence of
sunspots). The effect of cosmic rays on the upper atmosphere
depends on how strong the shielding from the Earth's magnetic
field is. The intensity of the geomagnetic field fluctuates by
about 50% in time periods of about 104 years. Another problem is the anthropogenic
influence (human civilization activity) on the distribution
of radionuclides in the atmosphere. It is both the burning of
fossil fuels that affect the CO 2
content in the atmosphere, and the radiocarbon released into the
atmosphere by nuclear tests, especially in the 50s. Determining
the concentration of radiocarbon is thus also of considerable ecological
importance. For accurate radiocarbon chronometry, all these
influences are distorting, it is necessary to take a correction,
compile a calibration curve by comparison with other
independent methods (eg with dendrochronology
- dating using annual rings in the wood of old trees).
Chronology of natural events using 10Be
Another cosmogenic radionuclide with a long half-life is beryllium
10Be. It is formed in the upper layers of
the atmosphere from oxygen and nitrogen by nuclear reactions with
secondary neutrons ejected by high-energy cosmic rays. 10Be with a half-life T1/2 of 1.6x106
years by beta- -radioactivity
decays into the stable isotope 10B. It is used to investigate rock weathering, soil
formation and erosion, lake sediment chronology. It is
interesting to measure the concentration of the beryllium isotope
10Be in
different layers of glaciers as an indicator of solar
activity in ancient times - as a result of the interaction of
cosmic rays and the solar wind (the
modulation of cosmic rays by the solar wind is discussed in
§1.6, the passage "Stellar
- solar - "wind"").
Luminescent
dating - optical and thermoluminescent
,
is based on the "deposition" of electrons released
during long - term irradiation of materials with natural
radiation in metastable levels and their
subsequent release by heating or light irradiation of the sample
(see §2.2, passage "Thermoluminescence and photoluminescence
dosimetry", paragraph "Luminescent
archaeological dating").
Long-time minerals dating
Radiocarbon method is
not applicable for
dating inorganic materials
such as rocks, and also for very long periods of time over
millions or billions of years. Here,
under certain circumstances, the decays of other
long-term natural radioactive
isotopes can be used, such as the decay of potassium 40K to argon,
the decay of the radioactive isotope rubidium 87 to strontium, or
the decay of uranium and thorium to the final element of the
decay series - lead. Radioactive dating by determining the
relative proportion of daughter radiogenic decay
products with respect to the parent nuclides
makes it possible in geology to determine the absolute age
of rocks *), the duration of individual geological
epochs, the age of meteorites, the Earth, the Moon and the solar
system. However, the accuracy of these methods is, in addition to
the technical difficulties of determining small amounts of
nuclides, limited by the variety of samples found and their
previous destinies.
*) Age
of rocks
Under the age of minerals in mineralogy and
geology, the time interval from the time of mineral
formation by solidification and crystallization from
molten matter to the present (measurement times) is understood.
It is assumed that during solidification, the material
composition of the mineral is closed
("preserved"), which then does not change otherwise
than radioactive transformation.
Note: Due
to the small concentrations of the investigated
radionuclides and their long half-lives, a simple method of
measuring the radioactivity of samples cannot be used.
Determination of the concentration of analyzed nuclides is
performed by mass spectrometry (see §2.6 , section "Magnetic spectrometers").
Potassium
- argon method for determining the age of minerals
The K-Ar dating method in nuclear geochronology
uses the decay of natural (primordial) radioactive potassium 40K with a half-life of 1.26 billion
years to stable argon 40Ar.
Natural potassium consists of three isotopes:
39K (93.258% - stable), 40K
(0.0117% - radioactive) and 41K
(6.73% - stable). The radioactive isotope
40K
decays with a half-life of 1,28.109
years in two ways: b- (89.1%) 40K ® 40Ca
+ e- + n to calcium; b+ (10.9%) 40K
® 40Ar
+ e+ + n to argon. In a mineral
containing potassium from the moment of its formation in
crystalline form (and closure), the radioactive isotope of
potassium 40K decreases and a
stable isotope of argon 40Ar,
resulting from the conversion of 40K,
accumulates. The age of the mineral can be determined by
measuring the content of both isotopes. The 40K
content is determined by measuring the potassium content of the
mineral and the known proportion of the isotope 40K
in the potassium. To determine the content 40Ar,
the sample is heated in vacum to a temperature of about 2000 °C,
whereby argon gas is released from the crystal lattice and the 40Ar content is determined by mass
spectrometry.
A complementary and
refining method of the K-Ar dating method is the argon-argon
(40Ar /39Ar)
dating method. The mineral sample is first irradiated
with neutrons in a nuclear reactor, whereby a reaction
of 39K(n, p)39Ar
takes place on the nuclei of a stable isotope of potassium 39K. The irradiated sample is then
heated in vacum to release both argon isotopes and their content
is determined by mass spectrometry. The age of the mineral is
determined from the ratio of the contents of the two isotopes of
argon (since the amount of 39Ar
formed by the reaction with neutrons is proportional to the
potassium content of the mineral).
Rubidium-strontium
dating
is based on the beta-
-radioactive conversion of the natural primordial isotope rubidium-87
: 87Rb ® 87Sr + e- + n´
to strontium-87 with a half-life of 4,8.1010 years.
If the mineral contains at least a small amount of rubidium, this
very slow radioactive transformation causes the content of
natural rubidium 87Rb to
decrease and the content of radiogenic strontium 87Sr
to increase since the formation of the mineral by crystallization
from molten rock. The age of the mineral can be determined from
the measured ratio of both isotopes. A complication, however, is
that minerals may contain certain amounts of natural strontium
and thus of the 87Sr isotope of
non-radiogenic origin *). Therefore, in order to accurately
determine the age of the analyzed mineral, it is necessary to
correct the determined content of 87Sr
to the content of non-radiogenic 87Sr.
The determination of another isotope strontium - 86Sr
is used for this (again by mass spectrometry). On the basis of
the known ratio of 87Sr/86Sr isotope in natural
non-radiogennic strontium (in minerals containing no rubidium),
can then be from determined whole content of 87Sr,
to subtract the content of non-radiogenic 87Sr.
*) The nuclei of the 87Sr isotope of non-radiogenic origin were formed together
with other strontium isotopes during the nuclear synthesis of
elements in space (in supernovae), not by radioactive
transformations of 87Rb.
Dating
of rocks by decay of uranium and thorium to lead
This method U-Pb, Th-Pb is based on measuring the current amount
of the parent element X (uranium 235U,
238U or thorium 232Th)
and the end daughter element Y of the respective decay
series (lead 207Pb, 206Pb or 208Pb)
- see above "Natural decay series", Fig.1.4.1. From the
decay law of the relevant parent element X with a
half-life T1/2, we then obtain
for the resulting age t the relation:
t
= T1/2 . [1 + NX(t) / NY(t)]
,
where NX(t) is the current
amount of the parent element X and NY(t)
is the current amount of the daughter element Y (of the
relevant lead isotope).
A suitable mineral that serves as a "carrier" for
uranium or thorium is zirconium silicate ZrSiO4, which is commonly found in rocks of
volcanic origin. During the formation of a zirconium crystal,
uranium or thorium, but not lead, easily enters its crystal
lattice. Therefore, when dating, we can assume that all the lead
in the sample is of radiogenic origin - it was formed by
the decay of uranium or thorium. .............
..... concordant dating - below ...........
Dating
using decayed radionuclides
The above methods of radiometric dating require that a certain
amount of a small, but measurable portion of the initial parent
radionuclide. This can be a problem with the oldest
samples originating, for example, from the beginnings of
the formation of our solar system. In such a case, paradoxically,
dating with radioisotopes with a shorter half-life
that have already decayed ("died out") and are not
present in the sample, may help. However, their breakdown
products, stable daughter nuclides, are
present in the sample. By measuring the concentration of these
extinct radionuclide daughter products by mass spectrometry, the
relative age of the analyzed sample can then be determined. In
co-production with the isochronous technique, it is possible to
calibrate, for example, the U-Pb method and specify the
determination of absolute age.
This category
includes 129I - 129Xe
iodine-xenon chronometry. When the supernova
explosion occurs, among others, as well as large amounts of
radioactive iodine 129I is
created. The radionuclide iodine 129
I with a half-life of 15.7 million years (which is the current view seems long, but the
astronomical point of view is quite short ...) converts by beta- -radioactivity to the stable xenon
isotope 129Xe. In xenon isolated from stone
meteorites of chondrites, a higher
content of the isotope xenon 129Xe
was found, than corresponds to its usual
representation in natural xenon. Surplus 129Xe
in chondrites was creates by radioactive decay of the isotope 129I (it is radiogenic 129Xe), which was chondrite material enclosed
in the formation of a solid silicate composition after cooling
the hot gaseous nebula of supernova. In the protoplanetary stage
of our solar system, this occurred in the period between about
4.8-4.5. 109
years, before the formation of our planetary system.
In practical use,
iodine-xenon chronometry is a relatively complex isochronous
laboratory method. The samples are irradiated with neutrons in a
nuclear reactor, whereby the stable iodine 127I isotope is converted to radioactive 128I by neutron capture
and the subsequent beta conversion to stable xenon 128Xe. After
irradiation, the sample is heated and the released xenon is
analyzed in a mass spectrometer, comparing the content of 129-I
and 129-xenon. ......
Determining
the time of formation of meteorites
Meteorites, especially some stone types called chondrites,
are among the oldest bodies in our solar system. They were formed
when the protoplanetary disk cooled, before the formation of
planets (.....). ... link ..... Isochronous dating method
Simple dating methods can be adversely affected by ignorance and
variability of initial conditions (different concentrations of
parent and daughter nuclides) and the possibility of migration of
relevant nuclides between the analyzed material and the
environment. To eliminate these effects and refine
long-term radioisotope dating, the so-called isochronous
method was developed. This more complex (but very
elegant) method uses the collection of mineral samples from two
or better several different parts (components)
of the analyzed rock and performs isotope analysis using three
isotopes: not only the parent and daughter element, but
also the content of another isotope of the
daughter element.
Thus, we have in the analyzed
material the starting parent radioactive nuclide X , which
decays with a time T1/2 to a
stable daughter radiogenic nuclide Y´; the relevant daughter
element has yet another stable Y isotope (of
non-radiogenic origin). Because all isotopes of a daughter
elementY have the same chemical properties, the ratio of
these isotopes NY´ / NY in the whole analyzed rock is the
same. In contrast, the X isotope has different chemical
properties and the initial NX /
(NY´ + NY)
ratio is different for different minerals at the time of rock
formation. Due to the decay of the radioactive isotope X,
the relative proportions of NX /
NY and NY´
/ NY in different rock minerals
will be different after a sufficiently long time.
The amount of nuclide Y´
in the analyzed material consists of the original initial amount
NY´(0) and the nuclei formed by
the radioactive decay of the radionuclide X. From the law
of exponential decay of the parent radionuclide X follows
for the time increase of the concentration of the daughter stable
radoiogenic nuclide Y' the relation NY´(t) = NY´(0) + NX(t).[el.t
-1]
. Dividing this relationship by the amount NY
of another stable isotope Y
we get the equation for the nuclide concentration ratios :
[N
Y´ /
N Y ] (t)
= [ N Y´ / NY ] (0)
+ [N X / N Y ] (t) . [e l . t - 1] .
If we choose the time t as a parameter, it is in the
ratios of nuclide concentrations the linear equation
y = a.x + b, whose graph is a line called an
isochron (Greek isos = the same,
chronos = time - isochron is a line connecting on the graph
of a place with the same time occurrence of the displayed
phenomenon). The slope of this line tg
a º a =
e l . t - 1determines the time t (age), the
intersection point (intercept) with a vertical axis b = [NY' / NY](0)
specifies otherwise unknown initial ratio nuclides Y' and Y
.
Therefore, we
measure the current content of NX(t),
NY´(t), NY(t)
of these three isotopes in several samples and plot
the ratio NX / NY
on the horizontal axis and the ratio NY´
/ NY on vertical axis (Fig.1.4.3
left). Using the least squares method, we interpolate the linear
regression function - representing the line isochron.
If the dependence NY´ / NY is a linearly
increasing function of NX / NY (individual points "fit"
well on a straight line), this indicates a good
correlation between the measured loss of radioisotope X
and the measured increase of the daughter isotope Y´. The
regression line has the form y = a.x + b, where b
represents the original ratio of the elements NY´(0)
/ NY(0). The resulting age t
is then determined from the relation :
t
= (ln2 / T1/2 ) . [N
Y´ (t) / N Y
(t) - N Y´ (0) / N Y (0)] /
[N X (t) / N
Y (t)] ,
where T1/2 is the half-life of
the initial parent nuclide X . The initial ratio NY´(0) / N (0) is found from the
regression line.

| Fig.1.4.3.
Isochronous and concordant analysis in long-term
radiometric dating. |
| Left:
Linear regression function - isochron
for Rb/Sr dating - interpolated points [NX/NY, NY´/NY]
from several samples of the same rock. |
Right:
Concordia curve for
235U + 238U
dating, together with discordia (error
line) fited with measured values of samples of
the same rock. |
|
The great
advantage of the isochronous method is its independence
from the original isotope ratio of the daughter element: this
unknown original ratio is obtained from the regression line. The
original ratio of Y and Y´ isotopes may have been
very different in the past, and yet the same age
comes out. If the points in the isochron graph do not lie on a
straight line, this indicates some events that have
disrupted a closed rock system throughout history. The
resulting value of age will then not be accurate,
and from the regression analysis we can determine the mean
square error of the obtained average value.
The most commonly
used triplets of isotopes for the isochronous dating method are
listed in Table ( X- the default parent
radoionuclide, Y´ - its stable daughter
nuclide, Y - another stable isotope of this
daughter element) :
| X |
T 1/2
[years] |
Decay of radionuclide X ® Y´ |
Y´ |
Y |
| 87 Rb |
4,72.10 9 |
b - : 87
Rb ® 87
Sr + e - +
n ´ |
87 Sr |
86 Sr |
| 147
Sm |
1,06.10 11 |
a : 147
Sm ® 143
Nd + a |
143
Nd |
144
Nd |
| 176
Lu |
3.8.10 10 |
b - : 176
Lu ® 176
Hf + e - +
n ´ |
176
Hf |
177
Hf |
| 187
Re |
4.3.10 10 |
b - : 187
Re ® 187
Os + e - +
n ´ |
187
Pers |
186
Pers |
| 235 U |
7,04.10 8 |
a, b - : 235
U ® 7
a + 4 b + 207 Pb |
207
Pb |
204
Pb |
| 238 U |
4,5.10 9 |
a, b - : 238
U ® 8
a + 6 b + 206 Pb |
206
Pb |
204
Pb |
As mentioned above, the
reliability and accuracy of long-term dating techniques is, in
addition to technical difficulties, limited by the variety of
samples found and, in particular, by ignorance of their previous
destinies over millions or even billions of years in their
history. In exact archaeological, paleontological and geological
dating, therefore, great emphasis is placed on the agreement
of the results obtained by various independent methods
- we are talking about the so-called concordant dating
(lat. Concordantiae = agreement,
consent, conformity) .
Concordant radioisotope dating
In radioisotope dating, compliance can be analyzed using two
different radionuclides, mainly uranium 235U and 238U.
It is used here that two radioactive clocks
imaginarily, "ticking" inside the
sample: the faster "ticking" uranium-235 transforms
with a half-life of 700 million years, the slower clock is based
on uranium-238 with a half-life of about 4.5 billion years.
Comparing the results from both of these chronometers makes it
possible to refine the determination of the age of the sample.
Two isochronous
equations follow from the decay laws for both uranium :
[
206 Pb / 204 Pb ] (t)
= [ 206 Pb
/ 204 Pb ]
(0) + [ 238 U / 204 Pb ] (t) . [e l238U . t - 1] ,
[
207 Pb / 204 Pb ] (t)
= [ 207 Pb
/ 204 Pb ]
(0) + [ 235 U / 204 Pb ] (t) . [e l 235U .
t - 1] .
If from several measured samples of the same rock we get using
these equations two identical independent values of age t
- there was a concordance (consent), it is a
significant indication of accuracy of the dating
method.
From these two
isochronous equations we can also create a so-called concordance
diagram (Fig.1.4.3 on the right). Plot the ratio 207Pb/235U
on the horizontal axis (on a linear scale) and the ratio 206Pb/238U
*) on the vertical axis. We use the time t as a parameter:
for different values of t we graphically plot the points {[207Pb/235U](t)
; [206Pb/238U](t)}.
This creates a theoretical curve called a concordia,
each point of which gives a concordant value of age t and
the ratio of the concentrations of lead and uranium isotopes,
provided that no nuclides have entered or escaped from the
sample. The values of specific samples from the analyzed mineral
are then plotted in this graph. We get points that do not
generally lie in the concordia, because the assumption of closure
may not be met. Since all isotopes of uranium and isotopes of
lead have the same chemical properties, their possible leakage
from the material is the same and the measured values of the
uranium to lead ratio should lie on a straight line. Therefore,
we interpolate a straight line with the measured points - called discordia
(Latin inconsistency, disagreement
, discrepancy), whose
intersections with the concordia represent the initial time t0 crystallization of the material ("maximum
age") and time t´ ("minimum age") corresponding
to a possible metamorphosis event during the history of the
sample. The concordant diagram serves as an internal
check of the dating method, allowing to detect events
that violated the conditions of correct dating.
*) These are 207Pb and 206Pb radiogenic lead : the initiation of 207Pb (0) and 206Pb (0) is subtracted
from the total measured amount of these isotopes using the
isochronous method .
Determination of
the age of the Earth and the Solar system
Radiometric dating with the help of radionuclides with a very
long half-life has been significantly applied not only to
geological minerals, but also to determine the age of our planet
Earth and the entire solar system. Using only geological methods,
determining the age of the Earth is problematic, inaccurate,
basically impossible. Due to geological processes -
weathering, plate tectonics, volcanism, hydrothermal processes -
the chemical and physical composition of the rocks changed
significantly.
In the first half of the 20th century was
the main pioneer of radiometric dating of rocks, geochronology,
A.Holmes, who with the gradual refinement of uranium-lead methods
determined the age of rocks in the range of about 1.5-3 billion
years. His successor, C.C.Patterson, also used the uranium-lead
method on the age of meteorites from the early
periods after the formation of our planet. Meteorites as cosmic
bodies are more advantageous for dating because they have not
been altered by any geological processes. In 1965, Patterson
measured samples from meteorites found near the Barringer impact
crater in Arizona and reached an age value of 4.55 billion years.
These measurements were repeated many times, averaging 4.56
billion years (± 1% deviation).
So this is the value of the age of
the planet Earth and at the same time the whole Solar
system. All the material in the solar system was formed
at about the same time, the various chemicals, including
radioactive isotopes, formed together. This is shown by
measurements of different meteorites from different regions of
the Solar system - individual planets formed at virtually the
same time (from an astronomical point of
view).
On the formation of stars and planets and
their chemical evolution, see §4.1, part "Evolution
of stars", on primordial
cosmological nucleosystesis §5.4, part "Lepton
era. Initial nucleosynthesis" in
monograph "Gravity, black holes and space-time physics". For determining the ages of various
objects in the universe (stars, galaxies, the
entire universe), see §4.1, passage "Ages of
objects in the universe".
Production of artificial radionuclides
For the needs of current science and technology, industry and
healthcare, we are far from sufficient with a few radionuclides
of natural origin (natural radionuclides
uranium 235 and 238 are, however, the basis of fission nuclear
reactors and nuclear energy based on them).
So we have to make radionuclides artificially :

Fig.1.4.4. By bombarding the target nucleus
A with an accelerated elementary particles, the nuclear
reactions produces the resulting core B, which may
be raduoactive.
The resulting nucleus B is initially formed
usually in the excited state B', followed by
deexcitation of gamma photon emissions. This is usually not
important for the production of radionuclides, except in
situations where the excited nucleus is metastable with a longer
half-life.
In order to produce a radioactive
nucleus from a stable nucleus, it is necessary to change
the number of protons or neutrons so that the
equilibrium configuration is disturbed. As discussed in the
previous §1.3 "Nuclear reactions", we achieve this according to Fig.1.4.4 by bombarding
initial target nucleus A suitable
particles - protons or neutrons (or alpha-particles, deuterons,
occasionally heavier ions) that enter into the nucleus and bring
about appropriate changes there - nuclear reactions;
the resulting nucleus B is formed (mostly in the excited state B*, after
emission the gamma radiation then in the ground state), which is often radioactive.
Production equation
and yield
Thus, if we irradiate a target containing N atoms of the
initial irradiated element (nuclei A) with a
beam of particles (protons or other charged particles from a
cyclotron, or neutrons from a nuclear reactor) with intensity I
[number of particles/cm2 /sec.], the atoms of the resulting radionuclide (B)
will gradually accumulate in the target. During the irradiation
time t , the activity A(t) of the desired radionuclide B
in the target will be approximately given by the production
equation
A(t)
= I. N. s . (1 - e -l .t ) ,
where s [cm2] is the effective
cross section of a given nuclear reaction and l [s-1] is the decay
constant of the emerging radionuclide B (related to the
half-life by the relation l = 0.693/T1/2). The amount of activity produced is thus directly
proportional to the intensity of the irradiating beam I,
the amount of target substance and the effective cross section of
the reaction *). Initially, this amount is approximately
proportional also to the irradiation time t , but the
effect resulting from radioactive decay of the radionuclide,
expressed saturation factor (1 - e-l T), the increase in the number of generated nuclei
gradually slows down and after about 5-6 half-lives of the
resultant radionuclide B, the saturation state (approx. 98%) is
already achieved, the rate of production and disintegration is
balanced, the activity no longer increases during further
irradiation. The resulting activity of the produced radionuclide B is
then A = I.N.s .
*) These regularities apply under simplified assumptions of a
homogeneous and time-constant beam of radiation and a thin target
containing many times more starting atoms (A) than the
number of emerging nuclei (B).
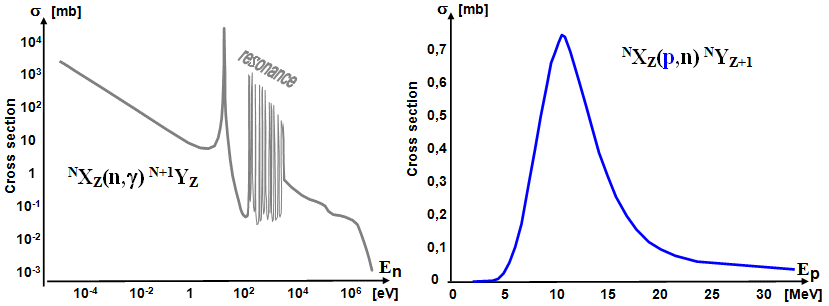
A typical dependence of the effective cross section of nuclear
reactions on the energy of bombarding with neutrons (left)
and protons (right).
The effective cross section of a
nuclear reaction, and thus the yield of
production, depends significantly on the energy
of the irradiating particles. This dependence is generally
complex and diverse for different types of production reactions.
When irradiated with neutrons,
the effective cross section for slow neutrons is
usually higher (although some reactions
take place on the contrary with higher energy neutrons). With increasing energy, the efficiency decreases
monotonically, as faster neutrons remain in the field of nuclear
forces for a shorter time, thus reducing the likelihood of a
nuclear reaction. In the region of tens and hundreds of eV, the
energy dependence appears to be a complex structure of resonant
maxima and minima (caused by discrete
levels of nucleon energies in the nuclei).
Irradiation with protons
(as well as deuterons or heavier positive particles) leads to
nuclear reactions only when a certain threshold energy
required to overcome the repulsive electrical (Coulomb) barrier
around the nucleus *) is reached, with the cooperation of the
tunneling phenomenon. Therefore, the effective cross section
initially increases significantly with energy, reaches a maximum
and then decreases monotonically (because
energy protons fly quickly through the region of the nucleus and
in this short time the nuclear reaction is less likely to take
place). For the simplest reaction (p, n) in
medium-heavy nuclei, the threshold energy of protons is about 5
MeV and a maximum yield is reached around 10 Me. For more complex
reactions, with alpha-particles and heavier nuclei, the yield
curve is shifted to higher energies, 20MeV and higher.
*) At lower energy, a proton can penetrate
the nucleus only through a quantum tunneling phenomenon
(§1.1, passage "Quantum tunneling
phenomenon"), with a much
lower probability.
These laws of energy
dependence apply not only to the required production reactions,
but also to possibly other parallel "parasitic"
reactions running in the target material, in which radionuclides other
than the required ones are formed - radionuclide
impurities. It is therefore necessary to set a certain optimal
energy, guaranteeing a high production yield with a
minimum content of radionuclide impurities.
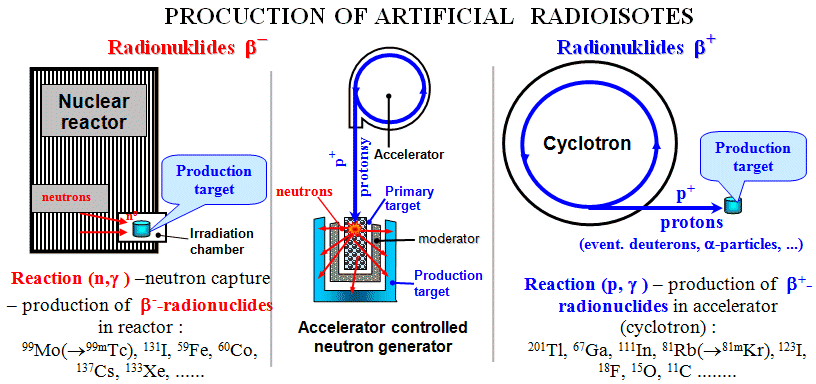
Fig.1.4.5. Production of radioisotopes by
irradiation of target nuclei in a nuclear reactor (left),
a neutron generator (middle) and a cyclotron (right).
Production
of radionuclides in a nuclear reactor
The easiest way is to irradiate the nuclei with neutrons
- since the neutron does not have an electric charge, it does not
cause electric repulsive forces and even a slow neutron willingly
enters the nucleus. The most common reaction here is simple neutron
capture (n, g) : NAZ + no ® N+1BZ + g *) , but reactions of type (n, p), (n, a) can also occur.
Irradiation with neutrons generally produces nuclei with an
excess of neutrons, which usually show radioactivity b-. The nuclear reactor is an intense
source of neutrons (§1.3, part "Nuclear reactors"), so that these b-
radionuclides are produced by irradiating a suitable target
material in the irradiation chamber of the reactor -
Fig.1.4.5 on the left. Some reactions of radionuclide production
by neutron irradiation are, for example: 6Li(n,a)3H,
14N(n,p)14C, 32S(n,p)32P, 98Mo(n,g)99Mo, .... Specific
methods for producing a number of important radioisotopes are
described below in the section "Properties of
some of the most important radioactive isotopes".
*) Note:
Neutron capture during the formation of radioactive nuclei is
also used in a very sensitive method of chemical composition
analysis - neutron activation analysis.
Irradiation of the examined sample with neutrons results in the
formation of radionuclides ("activation"), after which
therelevant radionuclide and the corresponding
(inactive) starting nuclide can be determined by
spectrometric analysis of the emitted radiation energies
(especially g) of the activated sample, using calibration also its
content in the investigated material. The method is
described in more detail in Chapter 3 "Application of
ionizing radiation", §3.4, section "Neutron activation analysis".
Another frequently
used method for the production of radionuclides in a nuclear
reactor is the irradiation of uranium 235U with neutrons, which causes the fission of
uranium nuclei into smaller nuclei that are radioactive,
eg :
235U + n ® 236U ® 131I + 102Y + 3n
® 137Cs + 97Y + 2n
® 133Xe + 101Sr + 2n
® 99Mo + 135Sn + 2n
® 155Sm + 78Zn + 3n
.........
and other radionuclides.
From these fission products,
the required radionuclides (e.g. 131I, 99Mo, 133Xe and others) are then isolated by
radiochemical methods. Since the heavy nuclei of uranium have a
significantly higher percentage of neutrons than the medium -
heavy nuclei formed by their fission, these radionuclides have an
excess of neutrons and show radioactivity b-.
A less commonly used
method for the production of radionuclides is their chemical separation
from fission products of uranium as fuel
in the reactor. In a nuclear reactor, 235U (or 238U) uranium nuclei split after the entry
of neutrons into two nuclei, chemically falling into the middle
part of Mendeleev's periodic table, which are mostly radioactive.
The most common nuclei formed in this way are, for example, 131 I, 137 Cs, 90 Sr, .... Spent fuel
cells from the reactor contain a large amount of these
radionuclides (of the order of TBq). However, it is very
difficult to radiochemically isolate individual radionuclides
from this diverse mixture so that the radionuclide obtained does
not contain traces of other radionuclides - in order to have a
satisfactory radionuclide purity, was not
contaminated.
Accelerator-controlled
neutron generator
In addition to nuclear reactors, accelerators
can also be intense sources of neutrons. Protons
accelerated to high energies are allowed to strike a (primary)
heavy metal target (lead, bismuth, tungsten),
where they cause fragmentation reactions in
which a number of particles and fragments, especially large
numbers of neutrons, are ejected from the target
nuclei. The neutrons slow down in the moderator (it can be water, which also cools the target) and hit the production targets, located
around the place of neutron formation, in which the nuclear
reactions produce the desired radioisotopes - Fig.1.4.5 in the
middle.
This method is still rather experimental,
but it seems relatively promising. In addition to the production
of radionuclides, it also enables the disposal and reprocessing
of radioactive waste with long-term radioisotopes and the
extraction of nuclear energy from thorium - it is discussed in
more detail in §1.3, section "Nuclear reactors",
section "Accelerator-controlled
transmutation technology ADTT".
Production
of radionuclides in the accelerator
For the production of positron b+ -radionuclides, on the contrary, it is necessary to add
protons to the nucleus. In order for the p+ proton to enter the
nucleus, it must be accelerated to a high energy of
the order of hundreds of keV to several MeV in order to overcome
the repulsive electrical Coulomb force of a positively charged
nucleus with its kinetic energy. The most common proton
accelerators are cyclotrons (§1.5, passage "Cyclotron"), which accelerate protons
by electromagnetic forces during many orbits in a circular path (maintained by a magnetic field) to
high energies. The proton beam is then led out from the circular
path by the magnetic field and impinges on a suitable target
material - Fig.1.4.5 on the right. Depending on the energy of the
protons, a number of reactions can take place. The simplest of
these is the radiation capture of a proton (p, g): NAZ + p+ ® N+1BZ+1 + g, but reactions of the type (p, p), (p, n), (p, d), (p, a) also occur,
especially at higher energies and in heavier nuclei.
For the purpose of the nuclear reaction
and transmutation, in addition to protons, the nuclei can be
irradiated with other fast charged particles: deuterons d
- take place mainly reactions (d, n), (d, p), a-particles
- reactions (a, p), (a, n) *) occur, or even heavier nuclei
or ions.
*) Note:
Nuclear reactions (a, n) are also used as neutron sources.
For this purpose need not have a helium nucleus artificially
accelerated, but suffice with suitable a -radionuklides
which we mix homogeneously with a suitable target material - some
light elements, which give a high yield of neutrons in reaction (a, n). The most
suitable is beryllium in the reaction 9Be(a, n)12C, which we mix with
a suitable a-radiator - used eg 210Po, 226Ra, 239Pu, 241Am. These mixtures are hermetically closed or sealed in
metal or glass containers and serve as portable laboratory
sources of neutrons, so-called neutron generators,
used eg in neutron activation analysis (§3.4 "Neutron
activation analysis").
Nuclei with an
excess of protons are mostly beta+ -radioactive or
decay by electron capture; according to the method of their
production, they are sometimes referred to as cyclotron
radionuclides. Some reactions of radionuclide production by proton
irradiation are, for example: 18O(p,n)18F, 13C(p,n)13N, 11B(p,n)11C, ....; deuterons eg 10B(d,n)11C, 56Fe(d,n)57Co, ...... Specific
methods for producing a number of important radioisotopes are
described below in the section "Properties of
some of the most important radioactive isotopes".
Acceleration of
negative ions - the
possibility of increasing the fluence power of the external beam
from the cyclotron
In the cyclotron is a standard accelerated heavy positively
charged particles - protons, deuterons, alpha particles,
or heavier nuclei such as carbon 12C. For small cyclotrons used for the production
of radioisotopes, an important requirement is the high
intensity - fluence power - of proton or deuteron beams.
At energies around 40MeV, a relatively large current in the beam
of about 2-5 mA is achieved. However, this maximum power can only
be fully utilized when irradiating an internal target
installed inside a vacuum accelerator tube. However, for the
routine production of radionuclides, it is more advantageous to bring
out the particle beam to irradiate the external
targets. In classical cyclotrons accelerating positive
particles, the resulting beam is extracted with an electrostatic
deflector. However, considerable dissipative heat
is generated at the partition of this deflector, which is a
limiting factor for achieving a high fluency performance of the
extracted beam.
This disadvantage is largely eliminated by
the technology of acceleration of negative ions (§1.5, part " Cyclotron ", passage "Acceleration of negative ions"). Hydrogen or deuterium
atoms are supplied with two electrons in an ion source in an
electric discharge, creating negative H- or D-
hydrogen ions with two electrons. These are then accelerated in
the cyclotron. After the necessary acceleration, a thin carbon foil
is inserted into the path of negative ions H- in the
appropriate path, which by "stripping"
removing their both light electrons and releases the desired
heavy p+
or d+.
This reverses the direction of curvature in the magnetic field,
causing their rapid brought out from the field of the cyclotron
to the external volume with corresponding
energy, with minimal thermal dissipation. This leads to
considerably higher performance production of
radionuclides in the outer target. This is especially important
for the production of larger radioisotope activities for
scintigraphic diagnostics (§4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy
') and a biologically-targeted radionuclide
therapy (§3.6"Radioisotope therapy") in nuclear medicine.
Modification
of irradiated material in the target. Radionuclide purity.
The basis for the production of the required radionuclide is :
¨ Selection of a suitable
production nuclear reaction
- the starting nuclide and the type of firing particles and their
energy, which implies the required irradiation equipment (nuclear
reactor, neutron generator, cyclotron) and its properties.
¨ Preparation of a
suitable target
of irradiated nuclide, its
chemical form and design. We can irradiate either the starting element
directly in elemental form or its suitable compound.
Isotopic enrichment of irradiated materials in
the target is often required - this increases the yield of the
reaction and facilitates the subsequent radiochemical separation.
Isotopic enrichment of target materials is technologically very
demanding and expensive. Therefore, after irradiation and
separation of the formed radionuclide, the remaining enriched
material is recycled for repeated irradiation.
The material to be irradiated in the
target is used in all three common states :
l Target in the solid phase
consists of a crystalline form, powder or amorphous material of
the starting target element or its compound. It is often a metallic
form of a given heavier element or its alloy. After
irradiation, the target material is used either directly
(encapsulated and a closed emitter is formed), or dissolves
in a suitable acid or hydroxide and performs radiochemical
separation and preparation of the required chemical form
of the radioisotope (or radioisotope labeling of a radiopharmaceutical
for nuclear medicine).
l A liquid target consists of an ampoule
with a starting element or its compound which is liquid at normal
temperature or is dissolved in a suitable solvent - most
often an aqueous solution. After irradiation, a
radiochemical separation is performed in the liquid preparation
and the resulting radioisotope is introduced into the required
chemical form or radiopharmaceutical.
l The gas target consists
of compressed gas enclosed in a suitable ampoule or chamber
(most often they are inert gases
xenon, argon, krypton, ...). At the end of
the irradiation, the radioisotope formed (contained in the
enclosure of the chamber or absorbed on the walls) is washed with
water or dilute acid; other radiochemical modifications are
analogous to the above targets. In the case of isotopically
enriched gas, it is recycled for reuse.
After irradiation, the target material contains not only
the desired radionuclide (at least never in 100% concentration),
but also a number of other atoms and possibly other radionuclides
formed by other (parallel, "parasitic") reactions. As a
rule, therefore, the irradiated material must be subjected to a
demanding procedure of radiochemical separation of
the required radionuclides.
Radionuclide purity ,
indicating the percentage ratio of the activity of the desired
radionuclide to the total activity of the preparation, is an
important parameter of the quality of the produced
radionuclide. For technological and medical applications, a high
radionuclide purity is generally required, better than
99.9% - or a radionuclide impurity content of
less than 0.1%. Radioinuclide impurities can be disruptive during
analytical or imaging measurements, or cause increased radiation
exposure of the patient (also discussed
§4.8, section "Radionuclides
and radiopharmaceuticals for scintigraphy", passage "Radionuclide purity of
radiopharmaceuticals").
Secondary radionuclides from decay products.
Radionuclide generators.
Some radionuclides are transformed into daughter nuclei, which
are not stable, but are again radioactive - they are secondary
radioisotopes. Obtaining these secondary radionuclides
from the decay products of other radionuclides can be an
efficient way to "produce" them.
This method is of particular importance
for short-lived radionuclides, which are formed
as daughter nuclei of radionuclides with a substantially longer
half-life. The relevant parent radioisotope, prepared by
irradiation at an accelerator or reactor, can be easily
transported to a remote laboratory, where a daughter short-lived
radionuclide can be separated from it
continuously or repeatedly, which is thus available for a much
longer period of time (given by the
half-life of the parent radionuclide). A
device that makes it possible to repeatedly separate a
short-lived radionuclide formed by the decay of another
longer-lived radionuclide is called a radionuclide
generator. Different physico-chemical properties make it
possible to separate the parent and daughter
elements in the generator. A generator is a system containing a
tightly bound parent radionuclide with a longer
half-life, from which the resulting daughter radionuclide with a
shorter half-life (which is no longer
firmly bound to the parent radionuclide carrier) can be separated (detach) either chemically (in
the liquid phase), hydrodynamically (elution) or by
blowing with air. The advantage of radionuclide generators is the
possibility to use appropriate short-term radionuclides even in
workplaces remote from the nuclear reactor or cyclotron. The
kinetics of radionuclide generators in terms of decay
equilibrium of parent and daughter radionuclides is
analyzed in §1.2, section "Mixtures of radionuclides, decay series,
radioactive balance".
A typical example of a radionuclide
generator is a molybdenum-technetium generator 99Mo/99mTc (described in more detail in §1.2, section "Gamma
radiation", passage "Radionuclide
generators", see also below
"Molybdenum-Technetium"), where beta-decay of
molybdenum 99Mo (T1/2 =
66 hours) produces metastable technetium 99mTc
(T1/2 = 6 hours),
which is a pure gamma emitter (Eg = 140keV) and has a wide
application in scintigraphy in nuclear medicine - see chapter 4 "Radionuclide scintigraphy", §4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy", where the principle of operation and technical
design of the 99Mo-99mTc generator is drawn in Fig.4.8.1. A rubidium-krypton generator used in ventilatory lung
scintigraphy is described below in the section "Rubidium-Krypton". In the
field of PET scintigraphy, the germanium-gallium
generator 68Ge/68Ga is finding an increasingly important application (see gallium Ga-68 below).
The table shows
several more frequently used radionuclide generators :
| Parent radionuclide
(T1/2 ) |
Type of decay |
Daughter
radionuclide (T1/2 ) |
Type of decay |
The resulting
nuclide |
| 68
Ge (275 d) |
EC ® |
68
Ga (1.14 hrs) |
b
+
, EC, g
® |
68
Zn |
| 81
Rb (4.7 hrs) |
b
+
, EC ® |
81m
Kr (13 sec) |
g
® |
81
Cr |
| 82 Sr
(25 days) |
EC ® |
82 Rb
(75 sec) |
b + , EC, g ® |
82 Cr |
| 90 Sr
(28 years) |
b -
® |
90 Y
(2.6 days) |
b -
® |
90 Zr |
| 99
Mo (2.78 days) |
b
- ® |
99m
Tc (6 hours) |
g
® |
99
Tc |
| 113
Sn (115 days) |
EC ® |
113m
In (1.66 hrs) |
g ® |
113
In |
| 188 W
(69.4 days) |
b -
® |
188
Re (16.98 hrs) |
b -
® |
188
Os |
| 227
Ac (21.77 years) |
a, b - series ® |
227
Th (18.7 days) ,
223 Ra (11.4 days) |
a, b -
series ® |
207
Pb |
| |
|
|
|
|
| |
|
|
|
|
"In vivo generators"
in nuclear medicine
In nuclear medicine, suitable compound of radionuclides -
the radiopharmaceutical - are applied to the internal
environment of the organism, where their enter into metabolic
processes, and by their biochemical pharmacokinetics
may accumulate in certain target tissues
and organs. If this radiopharmaceutical is labeled with a
radionuclide that converts not to a stable, but to
another radioactive isotope with a shorter half-life,
then after its uptake, another shorter radionuclide is
generated and accumulated in vivo
in the target tissue, which "cooperate"
with the primary parent radionuclide. If this in vivo
generated daughter radionuclide emits g-
photons or positrons, can be used for scintigraphic
imaging (planar, SPECT, PET - Chapter 4
"Radioisotope
scintigraphy"); if
emits alpha particles or beta-
(event. Auger electrons), can serve (or
contribute to) the radionuclide therapy - §3.6
"Radioisotope
therapy". This situation, when a longer-term
radionuclide applied to the organism, by its radioactive
transformation creates a daughter short-term radionuclide (or the
whole decay series) in the target tissue, is sometimes called an in
vivo radionuclide generator - in the context with the
above laboratory radionuclide generators.
A certain problem
with in vivo generators is the release - dissociation - of
daughter atoms from radiopharmaceutical molecules, leading
to impaired stability and redistribution of daughter
radionuclides. In addition to chemical processes - changes in the valence (oxidation number) of atoms
during radioactive transformation - the backscattering
of nuclei during radioactive emission of alpha,
beta and partially gamma quanta also contributes to this (§1.2, passage "Accompanying phenomena during
radioactivity", paragraph
"Backscattering cores").
The kinetic energy of the backscattering of nuclei usually exceeds
the binding energy many times the relevant atoms in the
chemical bond *). Radiochemical methods evolve labeling of
biomolecules, which penetrate inside the target cells and which
are internalized, while the daughter atoms may
be sufficiently long to keep in target cells.
*) This manifests itself mainly in
alpha-radioactivity. E.g. with an alpha particle emission of 4-7
MeV, a kinetic energy of approx. 60-100 keV is transmitted to the
daughter nucleus with a nucleon number of approx. 220 by back
reflection. The chemical binding energy of the respective atoms
in the biomolecules (conjugates, for example with monoclonal
antibodies) is only about 2-5 eV.
A typical example of
an in vivo generator used is radium 223Ra
which is applied as a radiopharmaceutical in the form of
chloride. It is taken up in bone metastases, in which 223-radium
decays "in vivo" with a half-life of 11.4 days by a
whole cascade of alpha-transformations (in
combination with an insignificant alpha-beta branch) to
other short-lived radionuclides: 223Ra(11,4d.; a) ® 219Rn(4s.; a) ® 215Po(1,8ms.; a) ® 211Pb(36,1min.; b-) ® 211Bi(2,2min.; a) ® 207Tl(4,8min.; b-) ® 207Pb(stab.)- see radium 223
Ra below for details. One complete radioactive transformation of the 223Ra nucleus in the
whole decay series to a stable 207Pb releases a total nuclear energy of almost 30 MeV,
mainly by four alpha-particles, with a high radiobiological
effect; gamma photons are also generated, which can be used to
gammagraphically monitoring the distribution of the
radiopharmaceutical. However, radium 223Ra does not allow chelating bonds to more complex
biomolecules such as monoclonal antibodies, so its use is very
limited ( 223Ra-chloride for palliative therapy of bone metastases).
A more
promising radionuclide type "in vivo generator"
is thorium 227Th,
which (unlike radium) allows using type
macrocyclic ligand DOTA chelating binding of 227Th with monoclonal
antibodies to form radioimmunoconjugates,
which are specifically sequester in tumor cells. High energy a particles capable
emitted in radioactive transformations, they can effectively destroy
these tumor cells - see thorium 227
Th below for details.
Another
radionuclide that could work very well as an in vivo
generator in targeted alpha-therapy is actinium 225Ac,
which decays with a half-life of 10 days by a cascade of 4 alpha
transformations (in combination with an
insignificant alpha-beta branch) to other short-lived
radionuclides: 225Ac(10d.; a) ® 221Fr(4,8m.; a) ® 217At(32ms.; a) ® 213Bi(46m.; b-) ® 213Po(4ms.; a) ® 209Pb(3,3h.; b-) ® 209Bi(stab.), is released energy about 27 MeV - see actinium 225
Ac below for details.
Here, too, the accompanying gamma radiation is generated, which
can be used for scintigraphic imaging. Radiopharmaceuticals
labeled with 225Ac (including monoclonal antibodies, eg 225Ac-Trastuzumab or 225Ac-PSMA-617)
are being tested for the treatment of leucemia, lymphomas,
neuroendocrine tumors, gliomas, melanomas, and are very promising
in the prostate. This "in vivo generator" method of
therapy proves to be significantly more effective than the
previously tested application of 213Bi alone (the disadvantage of
which is the short half-life of 46 minutes).
Therapeutic use of in vivo
radionuclide generators is discussed in more detail in
§3.6, passage "Alpha and beta radionuclides for therapy".
Chemical compounds of radionuclides.
Radioisotope marking. Radioactive preparations.
Radioactive nuclei - radionuclides - are normally part of the atoms
(which have the same chemical properties as
non-radioactive atoms) that make up the radioactive
substance. Only in exceptional cases is it a pure radioactive
element, mostly radioactive compounds of
own radioisotope atoms with other non-radioactive atoms, often in
a mixture with other substances. A radioactive substance
specifically prepared for laboratory, technical or medical use is
often called a radioactive preparation.
Natural
radionuclides (described above in the
section "Natural radionuclides") are dispersed in the
natural environment with a very low concentration. The heavy
elements uranium-238,235 and thorium-232 are contained in the earth's crust in
minerals in the form of oxides, silicates or phosphates.
Potassium K-40 is
abundant together with normal non-radioactive potassium. Tritium H-3 is in the form of "heavy" water 3H2O, carbon C-14 is in the atmosphere in the form of carbon dioxide 14CO2 . In plants,
radio-carbon and tritium are incorporated into virtually all biomolecules in cells, and later in animal and human
cells.
A special field of radiochemistry
deals with chemical reactions with the participation of
radioactive atoms. The targeted chemical attachment of the atoms
of a particular radionuclide to the molecules of a given
substance is called radioisotope labeling. The
labeled substance - radio-preparation - is then mostly
used as a radioindicator for various analytical
and diagnostic procedures (§3.5 "Radioisotope tracking methods" and §4.8 " Radionuclides and
radiopharmaceuticals for scintigraphy"), or as a radiotherapeutics
in biologically targeted radioisotope therapy (§3.6, section "Radioisotope therapy").
Chemical reactions of labeling with the
desired radionuclide generally do not proceed in 100% yield.
Therefore, in the resulting preparations, in addition to the
required radioactive substance itself, there is always a small
amount of unbound activity and possibly other radioactive
substances, that have different radiochemical properties and may
be disruptive impair the binding specificity of
the labeled substance in laboratory, diagnostic or therapeutic
applications. In addition to the radionuclide purity
discussed above, the radiochemical purity of the
preparations is therefore an important quality parameter. It is
the share of the declared chemical compound of a given
radionuclide in the total activity of the preparation.
Part of the
generated ionizing radiation is absorbed inside
material of the radioactive preparation, self-absorption
of radiation in the sample occurs. This absorbed
radiation can cause chemical changes in the material (§5.1, section "Effects of radiation on the substance") - radiolysis of
the labeled substance by its own radiation (most often beta or
alpha). During this radiation decomposition, the
required labeled substance decreases and other substances with
different properties are formed instead. This worsens the
radiochemical purity of the preparation. In particular,
radiolabeled complex organic molecules readily undergo
radiolysis. The higher the specific activity of the
preparation, the faster the radiation decomposition takes place.
Radionuclide
decay (transformation) schemes
The so-called transformation schemes are
used for a clear and comprehensive presentation of various types
of radioactive transformations and energy levels in specific
atomic nuclei (Fig.1.4.6). The parent and daughter nuclei are
represented in these diagrams by means of horizontal
lines (representing the energy levels of the nuclei),
the position of which in the diagram is determined as follows:
the proton number Z is on the horizontal
axis, the position in the vertical direction is given by the
nucleus energy E *). The basic energy
state of each nucleus is marked by a thick line,
the excited states of the nucleus are drawn in thin
commas (with data on energy and possibly other characteristics),
at the appropriate vertical height above the baseline. For the
basic states of radioactive nuclei, the half-life
is indicated, for special purposes event. also other
characteristics (eg spin). The basic state of a stable
core is sometimes drawn by a combination of a thick
solid line with hatching below. Metastable
energy levels are drawn by semi-bold dashes with
an indication of the lifetime (half-life) of this metastable
excited state.
*) In the practical drawing of decay
schemes, the exact proportions of energy values and proton
numbers are usually not strictly observed, only the relevant
relations are observed - states with higher energy are drawn more
above, nuclei with larger proton number Z are more to the
right of nuclei with smaller Z.
The radioactive alpha and
beta transformation of nuclei is to draw by an oblique
arrow to the left or right, connecting the parent and
daughter nuclei at their respective energy levels, which takes
place in a given process; next to this arrow is writen the type
of transformation (a, b, EC) and the corresponding quantum energy of the
radiation. Deexcitation of excited levels, ie isomeric
transitions g, are drawn by vertical arrows
connecting the higher levels with the corresponding resulting
lower levels, or with the baseline state of the daughter core.
For arrows showing nuclear transformations and dexcitation, their
relative representation [%] (probability,
intensity) is given - the average number of emitted
quanta (alpha, electrons or positrons, photons) per 100
transformations. At the bottom of the decay scheme (below the daughter nucleus line, or to save space in
another free space in the figure), the total
conversion energy Q [keV or MeV] is listed.
Close to the
horizontal lines showing the fundamental and excited energy
states of nuclei, their energies are given in
the decay diagrams. The values of this energy are determined
relative to the ground state of the daughter nucleus,
to which the energy "0" is assigned.
The basic state of the parent nucleus then has the energy Q
and possibly the excited states of the daughter nucleus have
correspondingly lower energies.
Branched
transformation
A more complex situation occurs in the case of branched
transformation, when a given parent nucleus is
transformed by two different types of radioactivity
(with a certain probability) into two different daughter
nuclei with different ground state energies
- see §1.2, passage "Mixed (combined) radioactivity -
branched transformations".
Then we have two different values of the total energy of
transformation Q and two series of energy levels of daughter
nuclei. For each branch of radioactive transformation, energies
are calculated independently. The ground state
of one daughter nucleus is assigned energy "0". The
same is done for the second daughter nucleus in the second branch
of the transformation. The parent nucleus then has two different
initial energy values, which we attribute to it from the side of
the diagram to which the type of transformation is directed. A
typical example of this situation can be seen in the decay scheme
of 186 Re , 192 Ir , 152 Eu and several others.
Figure 1.4.6 shows
some of the simplest typical decay schemes :
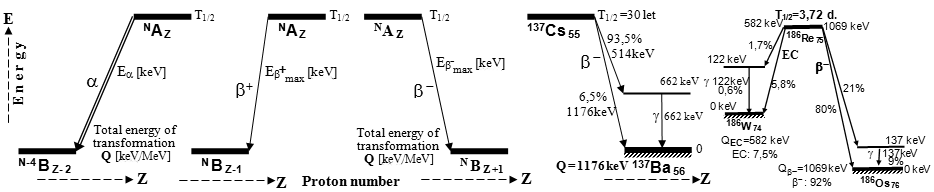
Fig.1.4.6. The simplest typical conversion schemes of
radioactivity a, b+, b-, b-+ g and branched transformations EC+ b- .
On the far left it is a pure decay a (mechanism
according to Fig.1.2.2 in §1.2), where
with the radiation of the particle a (helium nuclei 4He2) the parent nucleus NAZ transforms into the ground state of the daughter
nucleus N-4BZ-2 about lower energy;
the nucleus B is shifted to the left by two
places, as it corresponds to the proton number Z-2, and down
according to the energy difference. For the arrow (a double arrow
is used for the alpha) showing the transformation itself, the
type of transformation and the corresponding quantum energy of
the emitted radiation are indicated.
Next to it is the conversion scheme of pure
radioactivity b+ ( NAZ ® NBZ-1 + e + + n), where the daughter nucleus B is
shifted by one place to the left relative to the parent nucleus A,
which corresponds to a reduction of the proton number by 1.
Furthermore, Fig.1.4.6 shows the decay scheme of pure
radioactivity b- (NAZ ® NBZ+1 + e- + n´, according to Fig.1.2.3 in §1.2), where the parent
core A is transformed into the basic state of
the daughter core B, shifted by one place to the
right.
For simplicity, we have so far disregarded
the fact that the net conversion of a or b to the basal level of the
daughter nucleus occurs in only a small percentage of cases;
usually the daughter nucleus is formed in an excited state,
followed by deexcitation - combined radioactivity a+g or b+g. The penultimate
decay scheme in Figure 1.1.5 on the right represents an example
of the radioactivity b of a specific 137Cs nucleus, which with a half-life T1/2 = 30 years is transformed into
a daughter nucleus 137Ba, which is stable. Only about 6.5% of cases go to the
basal state of barium, while as many as 93.5% of cases go to the
excited state of the nucleus 137Ba with an energy of 662 keV, shown by a horizontal
comma. The vertical down arrow shows the deexcitation of this
excited state by emitting a photon of radiation g with this energy
662 keV (the properties of this important
radionuclide 137Cs are described in more detail below in the passage
"Cs-137").
On the far right of Fig. 1.4.6 is an
example of the above-mentioned branched radioactive
transformation of one parent nucleus by two
different types of radioactivity to two
different daughter nuclei (the
example is a simplified scheme of radinuclide 186Re, described in
more detail below in passage 186 Re ).
The decay patterns of some radionuclides
are quite complex, with a number of cascading transformations of
corpuscular and a number of excited energy levels, between which
isomeric transitions occur accompanied by gamma radiation quanta.
That correspond to the complex spectra of such
radionuclides. Spectrometric analysis of the emitted radiation is
then the main method of recognizing the structure of nuclides.
In the following section "Properties
of some of the most important radionuclides"
we present specific decay schemes of a number of more important
and more frequently used radionuclides, together with their
radiation spectra and other properties, including applications :
Properties
of some most important radioactive isotopes
Of the large number of radionuclides (now more than 2000 are
known), some of which occur in nature, but most are produced
artificially, only less than a tenth are important and practical.
Here we will get acquainted with some radioisotopes that are
particularly interesting or important from the point of view of
natural science or for practical
applications. We will describe these important
radionuclides in more detail with the indication of their
properties, decay schemes and radiation spectra *), methods of
origin or production and their use. In the introduction to the
description of the properties of individual radionuclides, we
always make a brief mention of the physico-chemical properties of
the relevant elements; although the radiation
behavior of the respective radionuclides is
not directly related to these properties (radioactivity is a property of the atomic nucleus, not
the electron shell), they are important for
radiochemistry of radioisotope preparation and their behavior and
distribution in nature, including living organisms, or
applications in medicine. In addition, many non-radioactive
isotopes are important in nuclear and radiation processes:
whether as a source of bombardment particles, as target
materials, components of detection media, shielding and
collimating materials. For a comprehensive understanding of
nuclear and radiation physics, it is useful to reflect all these
contexts...
*) Gamma
radiation spectrometry
of some used radionuclides we performed at our Department of
nuclear medicine in Ostrava-Poruba, within its modest
possibilities, in the 70's and 80's on a scintillation NaI
(Tl) detector (size 5x5cm) and a semiconductor Ge(Li)
detector with the help of a 4096-channel analyzer ICA-70,
later using a Canberra-Packard computer analyzer. We no longer
have a Ge(Li) detector at our workplace (ended
service...) and the semiconductor spectra
below were acquired at the HPGe detector of the
spectrometric laboratory of the SÚJB and SÚRO regional center
in Ostrava-Zábøeh with the kind cooperation of colleagues Ing.
J.Lušnák and Ing. J.Rada- the
author thanks them very much !
The spectra of soft gamma radiation
and characteristic X-rays (with
energies lower than 30 keV, for which the detection efficiency of
the coaxial high-volume HPGe detector is already very low) were measured separately on a planar semiconductor
detector with a beryllium input window.
Some of these measurements were mostly
performed many years ago on analog spectrometers, so we built
them into our spectra by scanning and interpolating the original
graphs of the spectra - we apologize for event. distortions and
degraded quality.
Many years ago, we tried the
beta radiation spectrometry very improvised with
the help of plastic scintillators and liquid
scintillators, which is definitely not optimal (unfortunately we never owned a magnetic spectrometer
...). We do not mention continuous spectra
of beta radiation for individual radionuclides in our treatise
also because they are quite "dull" and uninteresting -
visually they are similar to "eggs like eggs", only
stretched to higher or shrunk to lower maximum energies. Relevant
information from them can be obtained only by demanding computer
analysis using the Fermi-Kurie graph method (§2.6 "Measurement
of beta, proton and neutron radiation. Liquid scintillators."; at that time we calculated it manually and
plotted it graphically on graph paper...). Conversion
and Auger electrons with discrete spectra of characteristic
energies would be interesting, but at our workplace we didn't
have an instrument to measuring...
Scintillation and semiconductor detector used in gamma-spectrometry
of radionuclides.
Left: Scintillation probe - NaI (Tl)
scintillation crystal + photomultiplier with shielding. Middle:
Analog-to-digital converter (ADC) and computer (CPU) multichannel
analyzer. Right: Semiconductor
Ge(Li)/HPGe detector with preamplifier and Dewar vessel with
cooling liquid nitrogen.
Common features of
gamma-spectra of radionuclides
Despite all the differences between the energy and the intensity
of gamma rays emitted by different radionuclides, the
gamma-spectra of all radionuclides have some similar features :
¨In
the beginning photon spectrum in low energy are often seen the
peaks of the characteristic X-rays, emitted at
electron jumps between the inner shells in the excited atomic
shells of the daughter element. These excitations of atoms during
radioactive transformations occur in two ways :
1. Internal conversion of gamma radiation (§1.2., passage "Internal conversion of gamma
radiation and X"), when electrons from higher energy levels in the atomic
shell jump to the vacancies after conversion electrons.
Particularly g -fotons low energy (units or tens of keV) to have a high
conversion ratio. Also, some isomeric transitions, highly
forbidden due to the spin "mismatch" between the
excitation and the ground state of the nucleus (an example is the
isomeric 131m Xe), are realized by an internal conversion mechanism.
2. Electron capture
(§1.2, passage "Electron capture (EC)"), in which electrons from
higher levels of the atom immediately jump to the vacancies after
the captured electrons (usually in the K shell). Thus, especially
with radionuclides transformed by electron capture, that the
characteristic X-rays are highly represented or even dominant
(see eg 125 I).
In our gamma spectra, measured by
scintillation NaI(Tl) and semiconductor HPGe detector, peaks Ka,b of characteristic
X-rays are displayed only for medium and heavy radionuclides (for some lighter radionuclides we measured low-energy
peaks of characteristic X-rays separately on silicon
semiconductor detector as mentioned above).
We will present and analyze the characteristic X-rays in our
spectra, only if it is significant - it is clearly visible in the
spectra and has a radiation significance.
¨ If a radionuclide emits more
gamma-ray energies, the strongest gamma-lines are usually in the
lower and medium energies (tens to hundreds of keV), while higher
energies are usually represented with significantly lower
intensity (see "Magnified sections of spectra"
below).
Magnified sections of spectra
In our basic spectra are displayed the most important nuclear
levels and radiation energies of a given radionuclide, which are
used in applications and are measurable by commonly available
detection techniques. The peaks of the weakly represented gamma
radiation energies are lost in the spectra graphically displayed
on a normal scale (given by the height of the most intense peak)
in the region around zero values. To display them, we therefore
use an enlarged section of the spectrum, shifted
above the horizontal axis; while the energies correspond to the
basic scale on the horizontal axis. These magnified sections are
marked with a magnification sign "< ", an up arrow and a magnification
multiple - eg " < á 16 x
"- slight magnification, or
" << á 256 x
"- strong magnification. This situation often occurs when
the radioactive transformation occurs to a larger number of
excited states of the daughter nucleus. Then it is usually more
likely to lower energy levels than to higher
excited states - the gamma spectra are represented with a higher
intensity peaks at lower energy as the high energy peaks are much
weaker, are often visible only at high magnification and long
acquisition time. This is just shown by the magnified sections of
the spectra.
Differences in
scintillation and semiconductor gamma spectra
If we look at the images below gamma spectra of a number of
radionuclides, we can at a glance see some significant
differences between spectra measured by scintillation and
semiconductor detector :
¨
Photopeaks for scintillation spectrum are rounded
and soft, as if "blurred" - energy resolution
is relatively imperfect (approx. 10% for
test line 662keV 137Cs) , close gamma-lines merge into
one photopeak. The semiconductor spectrum
consists of very sharp and narrow peaks - the
energy resolution is about 30 times better. Some compact peaks
from the scintillation spectrum are decomposed into several gamma
lines on the semiconductor spectrum...
¨
In scintillation spectrum shows a
significantly represented continuous component of
Compton scattered radiation, especially in the region of lower
energies. This continuous background is disturbing (especially in the "peak" region of the
backscatter, which can interfere with the actual gamma peak of
the radionuclide being measured). In semiconductor
spectra, the continuous component is strongly suppressed,
because better energy resolution leads to narrow and high
peaks (while maintaining the same
area under the peak), which automatically
leads to a reduction in the relative height of the continuous
background on a graphical representation of the spectrum,
normalized to the maximum of the peak.
¨
We can observe some differences in the relative
intensity of the peaks, which are caused by different
energy dependence of the detection efficiency. The scintillation
NaI(Tl) detector also measures the gamma line with high
efficiency with energies of about 20-40keV, for which the coaxial
HPGe detector (used by us) already has a very low (or zero end)
sensitivity. Therefore, for example, characteristic X-rays
usually have a significantly lower content on the semiconductor
spectrum than on scintillation. For lighter radionuclides, X-ray
peaks do not appear at all on this detector. A planar Ge(Li) or
Si detector was used to measure them in some spectra.
Author's apology - distortion in the display of
spectra
The original measured spectra of radionuclides from a
multichannel analyzer (with high energy resolution and a number
of details) had to be significantly reduced graphically
for capacity reasons to display in our treatise. This often led
to geometric distortion of the spectra, especially from
semiconductor detectors - for example, to "shrinkage"
of the lines of photopeaks, loss of some points in the curves and
their disconnection, merging of nearby lines. As far as possible,
I tried to "retouch" these distortions and liken the
displayed graphs to the original spectrum. However, this has not
always been completely successful, I apologize and welcome
the comments of colleagues - experts of spectra ...
The same applies to the display of more
complex decay schemes of some radionuclides, where the
plotting of nearby energy levels and arrows of transformations
and transitions can also overlap and merge... For similar
reasons, the energy scales below the horizontal axes of
the spectra are only approximate (we are
able to ensure the accuracy of displaying the positions of
numerical values not better than about ± 10%).
For our purposes, however, this does not matter, because the
exact values of the energies of the individual peaks, read from
the original spectra (uncompressed, in digital form on the
spectrometer), are explicitly descripted by the arrows denoting
the respective photo peaks.
The scale on the vertical axes of the
measured number of pulses is relative, normalized to the
value of 100.103 pulses.
The individual
radionuclides are basically listed in the order of proton
and nucleon numbers, but with a number of minor
exceptions - on the one hand according to the sequence of
radioactive transformations (eg radium-223
is listed after radium-226, from which it is prepared), on the second hand, according to the importance and
method of use of the respective radioactive isotopes (e.g. cobalt 57-Co is mentioned up to the far more
important 60-Co). If, for a certain
element, its radioactive isotope do not "deserve" a
separate section by their number or significance, two or three
close elements are combined into one passage (eg nitrogen + oxygen + fluorine, chromium + iron,
copper + gallium + germanium, transurans) and
their isotopes are discussed in mutual context. This is logical
especially in the case of continuity of parent-daughter
radionuclide relationships (eg
rubidium-krypton, molybdenum-technetium, decay series of heavy
alpha-radionuclides).
Diversity of
radionuclides
Every radionuclide is certainly something interesting. However,
describe in detail the properties of all known
radioisotopes would be too lengthy and confusing *) (and it would certainly go beyond the powers of the
author...). Therefore, we do not
mention here those radioisotopes that were once or
several times, rather accidentally, in the context of the time,
used in research of some natural processes, material and
biological, or in nuclear medicine, and then no longer used -
either because they have their one-off role already
fulfilled, or have been replaced in
practice by more advantageous radionuclides
(which we therefore present). We analyze in more detail
radionuclides of particular interest from a nuclear and general
natural-scientific point of view, or widely used in scientific
and technical applications, medicine, industry (especially those that the author personally uses in
analytical and measuring methods, or as calibration standards). This choice may be somewhat subjective
- I welcome the comments and suggestions of colleagues..!..
*) It is not important to deal with, for
example, extremely short-lived radionuclides
that can not be used in any way - can be interesting at most in
terms of nuclear physics, studies of strongly nonequilibrium
nuclear configurations.
For individual radionuclides, we present
and display in the spectra mainly relevant
energy levels and radiation, which can be detected by available
radiometric techniques. Radiation intensity is
given numerically in percentages (eg "gamma 320keV (10%)" means that "10
gamma photons with an energy of 320keV are emitted per 100
conversions of the respective radionuclide"). A large number of other, low-saturated levels and
very poorly represented radiation energies, as well as their
other characteristics (such as spin,
multipolarity, parity, which are important only for nuclear
research) can be found in detailed isotope
decay tables. At our workplace, we mainly used the book edition
of the "Table of isotopes" by Lederer,
Hollander, Perlman (the new electronic
version is "http://ie.lbl.gov/toipdf/toi20.pdf"). Other
new detailed tables in the electronic version are "http://ie.lbl.gov/toi/nucSearch. Lund univ.") and "http://www.nucleide.org" from "Henri Becquerel Laboratory".
Short-term and
long-term radionuclides
One of the main factors determining the importance and use of
radionuclides is the half-life. As mentioned
above in section "Natural
radionuclides", these natural
radionuclides (primordial and cosmogenic) have very long
half-lives. Also, the vast majority of significant artificial
radionuclides used in science and technology or industry, have a
sufficiently long half-life - months, years,
decades or more, which allows their long-term use, especially in
the form of closed radiators.
Exception are some short-term
radionuclides used in nuclear medicine,
which due to their chemical and pharmacokinetic properties find
application in radionuclide diagnostics or therapy in the form of
open emitters - labeled radiopharmaceuticals,
applied directly to the body (usually
intravenously or orally, see Chapter 4 "Scintigraphy", §4.9 "Clinical
scintigraphic diagnostics in nuclear medicine"). In this case, the short
half-life may be an advantage in terms of the
radiation exposure of the organism.
Such short - lived radionuclides are
mainly technetium 99m
Tc (T1/2= 6 hours) and also light positron
radionuclides: carbon 11C (T1/2 = 20.4 min.), nitrogen 13N (T1/2 = 10 min.), oxygen 15O (T1/2 = 122 sec.) and especially fluorine 18F
(T1/2 = 110min.),
which is taken up in the form of 18F-deoxyglucose and accumulates especially in tumor
tissues, which are then imaged by positron
emission tomography based on the coincident detection of
gamma 511keV annihilation radiation
(see § 4.3 "Tomographic
cameras" and §4.8 "Radionuclides
and radiopharmaceuticals for scintigraphy", part "Positron radionuclides"). Their properties, decay schemes and the spectrum of
511 keV are shown below (passage "oxygen, fluorine"). The shortest radionuclide
used in nuclear medicine is metastable krypton 81
m Kr with a half-life only 13.1 seconds.
 Hydrogen
Hydrogen
The lightest and most widespread element in
nature (in universe) is hydrogen
H1 (hydrogenum
- water-forming),
whose proton nuclei formed just after the Big
Bang in the hadron era of the universe (§1.1, part "Cosmic
nucleogenesis"); accounts for 75% of total mass (baryonic) in the universe. It is the main
"fuel" of thermonuclear fusion inside stars ("Gravity and evolution of stars", part of the "Thermonuclear reaction inside
stars"). It is
very reactive, so in terrestrial nature it occurs practically
only in compounds (elemental hydrogen
occurs only in natural gas and volcanic gases), of which the most important is water H2O. Along with carbon, hydrogen is
the most important biogenic element. It has three important isotopes
(in total, the isotopes 1H1 to 7H1
are known). Basic
"light" hydrogen 1H1 (called sometimes protium)
- relative
representation of 99.9885 %,
"heavy" hydrogen, deuterium, 2H1
(0.0115%) and "extra heavy" hydrogen tritium 3H1
(cosmogenic nuclide with trace representation), which is already
radioactive.
Hydrogen 1
H ,
as already mentioned, is the main "fuel" nuclear
fusion inside stars. In nuclear technologies, hydrogen nuclei - protons are main shelling particles in
nuclear research, in the preparation of artificial radioisotopes (see above "Production
of artificial radionuclides"), in proton radiotherapy (§3.6, section "Hadron
radiotherapy").
Deuterium 2
H
- a heavier isotope of hydrogen, formed just after the beginning
of the universe during primordial
nucleosynthesis (it is analyzed in more
detail in the section "Lepton
era. Initial cosmological nucleosynthesis" §5.4 monograph "Gravity,
black holes and space-time physics"). Deuterium
is another non-radioactive isotope important in nuclear and
radiation physics. Deuterium nuclei deuterons d = 2H are often used as shelling particles in cyclotrons.
Heavy water is sometimes used as a moderator and coolant in
fission nuclear reactors (§1.3, section
"Nuclear reactors"). Together with tritium,
deuterium is most promising fuel for controlled
thermonuclear fusion (§1.3,
section "Merging nuclei"); direct
nuclear fusion of hydrogen nuclei 1H = proton in terrestrial conditions we cannot yet
perform.
Tritium
3 H
Leaving aside the free neutron (which is b- -radioactive no ® p+ + e- + n´ with a half-life
of » 13
min., max. energy beta 782keV), the lightest radionuclide is the
hydrogen isotope tritium 3H, which is with a half-life of 12.3
years converts by b- -radioactivity to the basic state of
the helium isotope 3He: 3H1 ® 3He2 + e- + n´ (its simple decay diagram is on the left in Fig.1.4.7) . Tritium is a pure beta emitter with a
relatively low maximum energy of emitted electrons of 18.6 keV (red curve in Fig.1.4.7 on the right).
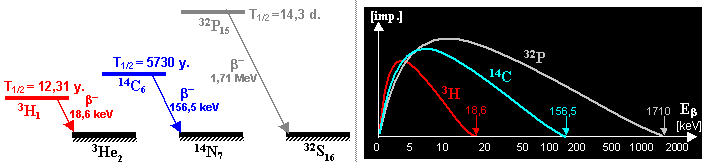
Fig.1.4.7. Beta - - radioactivity of some simplest isotopes.
Left: Conversion schemes of tritium 3H, carbon 14C and phosphorus 32P. Right:
Continuous beta spectra of these radionuclides.
Note 1: The
energy axis is nonlinear and irregular (illustrative only) in
order to draw very energetically different spectra .
Note 2: The spectra were measured with
a MarkIII Nuclear Chicago instrument with a toluene liquid
scintillator (§.2.6, section "Liquid scintillators",
Fig.2.6.2), plotted and (with modifications) interpolated.
Note 3: The shape of the low-energy
part of the spectrum (smooth growth from zero values - which is
somewhat different from the actual shape of the initial part of
the spectrum b- - cf.
§1.2, passage "Difference of energy spectrum b - and b + ", spectrum in the
left part of the picture) is affected by low sensitivity of the
instrument for low beta energy and electronic signal filtering to
reduce noise pulses.
In terrestrial nature, tritium occurs as a cosmogenic
radionuclide (see above the
section "Natural radionuclides",
or §1.6, part "Cosmic radiation", passage "Secondary cosmic radiation"). It also arises during the so-called ternary
fission of heavy nuclei, especially uranium (§1.3, section "Fission
of atomic nuclei"). The tritium content in nature has been significantly
affected by human activity in the last few decades. A large
increase in the content of 3H in the environment occured in 1960s as a result of
tests of thermonuclear weapons in the atmosphere (at thermonuclear explosion of lithium deuteride 6Li2H the tritium is
formed by the reaction 6Li (n, a) 3H - see §1.3, section "Compounding
of atomic nuclei", passage "Explosive
thermonuclear reactions"). In
fission nuclear reactors, tritium is formed in circulating
cooling water by the reaction of neutrons with deuterium 2H (n, g) 3H; to a small extent
with deuterium contained in ordinary water, more effectively when
using heavy water as a moderator and coolant. Furthermore, in
cooling water containing boric acid to control the reactivity of
the fission reactor, tritium is formed by the reaction of 10B (n, 2a) 3H. During the
operation of nuclear power plants, therefore, a certain smaller
amount of 3H
(in the order of hundredths of cosmogenic
tritium) enters the water and gas
discharges.
Tritium is a suitable excitation
radionuclide in small electrical sources - beta
- voltaic cells - see §1.3, passage "Radionuclide volta cells" ("atomic batteries").
For many applications in nuclear physics (especially for thermonuclear fusion,
in the future probably for energy use - §1.3 "Nuclear
reactions and nuclear energy", part "Fusion of atomic
nuclei"), in biology and medicine, tritium is produced
artificially by reacting neutrons with lithium: 6Li (n, a) 3H, or is obtained from
heavy water in reactors.
Helium
The second lightest and most widespread element in the universe, helium
He2 ("sun god element"), formed in the Lepton
era after the Big Bang (primordial nucleosynthesis is
analyzed in more detail in the "Lepton
Era. Primary Cosmological Nucleosynthesis" section in §5.4 of the monograph "Gravity,
Black Holes and space - time physics").
However, it is rare on Earth - the reasons are given in §1.1,
section "Cosmic nucleogenesis". All helium in terrestrial nature is a product of
the radioactive alpha-decay of uranium and thorium. Under normal
conditions, helium is a chemically inert gas
which, due to the very low liquefaction temperature (-269.9 oC = 4.2 oK) has an important
application in cryogenic technology, especially
in superconducting electromagnets. It has two stable
isotopes: 4He (98.999863%) and 3He
(0.000137%). Helium-4 is a very strongly bound nucleus, as it has
both the proton and neutron shells filled, which are the lowest.
Radioactive isotopes of helium have no practical significance, as
they are very short-lived *). Of the 7 known
radioisotopes of helium, 5 He is the "most stable" (formed by
irradiation of beryllium with neutrons: 9Be4 + 1n0 ® 6He2 + 4He2), which has a half-life of only 0.81sec! However, in
nuclear physics, helium (4He) is important as an efficient cooling medium
(superconducting electromagnets of accelerators and tokamaks,
cooling of sensitive detectors) and also as a product of
thermonuclear hydrogen fusion (§1.3 "Nuclear reactions and nuclear
energy", part "Fusion of atomic
nuclei"). Helium nuclei - alpha particles - are
the essence of alpha-radioactivity.
*) Diproton: the short-term isotope of helium 2He2
, also called diproton 2p, has a certain
interest in nuclear physics - consists of two protons, without
neutrons. This state is very unstable and breaks down almost
immediately by proton emission or beta+ -decay. A separate diproton was not observed, only in
highly excited states, eg 18Ne, the simultaneous emission of two protons was
observed, which was interpreted as originating from the
short-term bound state 2p.
Lithium,
Berylium, Boron
Elements of this group, despite being very light, are relatively under-represented
in nature for astrophysical reasons. They formed, as primordial
elements, in small quantities in a rapidly expanding
universe at the end of the Lepton era ("Lepton era. Initial cosmological
nucleosynthesis") and in later stellar nucleosynthesis they no longer
form, but are burned (§1.1, part "Cosmic
Alchemy - we are
descendants of stars! , passage "Representation
of elements in nature" and §4.1 "The
role of gravity in the formation and evolution of stars",
part "Evolution of stars" in book "Gravity,
black holes, and physics of space-time").
Lithium Li3
is a very soft light metal from the alkaline group (it is the lightest metal, and the same time the
lightest element in a solid state at room temperature), chemically highly reactive, so they occur in nature
only in compounds. It has two stable isotopes 6Li (7.5%) and 7Li (92.5%). Although
lithium has no significant longer -lived radionuclides (the longest half-life 0.84 s. has 8Li), this element is important in
nuclear physics. 6 Li serves as a neutron absorber and source material for
tritium production in controlled thermonuclear
fusion (§1.3, part "Tokamak"). A mixture of lithium and
deuterium - lithium-6 deuteride 6Li2H was misused as fuel for the "hydrogen" thermonuclear
bomb (§1.3, part "Fusion of atomic
nuclei", passage "Thermonuclear
explosion").
Beryllium Be4
is a hard gray metal that oxidizes easily in moisture; it is rare
in nature (approximately 3-10 mg/kg in the earth's crust) in
compounds such as emerald. It is used in special alloys
in metallurgy. In nuclear physics, its low absorption
(high transmittance) is used for low-energy X and gamma radiation
- beryllium entrance windows of sensitive detectors.
From beryllium is also fabricated some sections of tubes in
accelerators, in which there is interaction of the particles - to
make the flying out particles easier (without
significant absorption) to penetrate to the
detectors. In a mixture with a-radionuklides, beryllium serves as a laboratory source
of neutrons (see, e.g. passage "Neutron
radiation and its interaction" in
§1.6). Beryllium has the only one stable
isotope 9Be. Of the radioactive isotopes of
beryllium, two cosmogenic radionuclides are of some scientific
importance :
Beryllium 7Be
, with a half-life of 53.3 days, is converted by electron
capture to a stable isotope 7Li - in 89.5% to the ground state, in 10.5% to the
excited level of 478keV, during the deexcitation of which gamma
radiation of this energy is emitted. The 7Be isotope is formed in nature by cosmic rays in the
atmosphere. These are mainly fission reactions of high-energy
protons with nitrogen nuclei 14N (p, 2a) 7Be or oxygen 16O (p, 10B) 7Be, then nuclear reactions of neutrons with nitrogen
nuclei 14N
(n, 8Li) 7Be and oxygen 16O (n, 10B) 7Be. Its detection is
used to study transport processes in the atmosphere. For research
purposes in nuclear physics, the 7Be isotope is artificially prepared by irradiating
lithium with accelerated protons in a cyclotron by the reaction 7Li (p, n) 7Be. The isotope 7Be is interesting for
nuclear physics in that it first proved the dependence of the
rate of radioactive transformation by electron capture on the
chemical state of the isotope - see §1.2, section "Electron capture",
passage "Two peculiarities of EC".
Beryllium 10Be
is transformed into a stable isotope 10B by b-
radioactivity with a half-life of 1.6.106 years in the basic
state. In the atmosphere, the action of cosmic radiation creates
reactions with released neutrons: 14N (n, p a) 10Be. Atoms of 10Be are trapped in the atmosphere on aerosol particles
and with rainwater reach the earth's surface, where they settle
in the soil, oceans and glaciers. Measurement of this 10Be deposition (by
mass spectrometry), due to its long half-life, is used to monitor
some geological and oceanographic processes.
A very hidden
(deep inside massive stars) natural
important radionuclide is one very short-lived isotope of
beryllium :
Beryllium 8Be , due to its extremely short
half-life 6.7.10-17 seconds, is completely insignificant from the point of
view of "terrestrial" nuclear physics. Almost
immediately after its formation, beryllium-8 breaks down into 2
alpha particles (helium nuclei). However, it is very important
from the point of view of nuclear astrophysics:
via Be-8, nuclear "helium" burns to carbon in the late
stages of massive star evolution. In the specific conditions of
high temperatures and pressures inside helium-rich massive stars
(after "burning out" of hydrogen), even such a short
lifetime of 8Be is enough for the capture of 4He nuclei in 8Be to produce a large amount of carbon 12C - it is discussed
in §4.1 "The role of gravity in the formation and
evolution of stars", part "Evolution
of stars" passage "Combustion
of helium" book "Gravity,
black holes and space-time physics".
Boron B 5
is a semi-metallic element found only in compounds in terrestrial
nature and has two stable isotopes:
10B (19.9%)
and 11B (80.1%). Although boron has no useful
radionuclides (all are very short-lived,
the longest 0.77s. has 8B), this element is very important
in nuclear technology. The high effective cross
section of the isotope 10B for the absorption of slow neutrons makes it very
suitable material for control rods in nuclear reactors,
or the addition of boric acid into cooling water for
reactivity control (§1.3, section "Nuclear reactors").
Carbon
Carbon C6 (Carboneum) is a very important element in the universe, where it is
formed by the thermonuclear fusion of helium in the interior of
massive stars (§1.1, passage "We are
descendants of stars! ",
in more detail "Thermonuclear fusion in the interior of
stars"). In massive stars of the 2nd
and subsequent generations, carbon also participates in the
thermonuclear fusion of hydrogen into helium (as a "catalyst" - CNO cycle). Carbon is the fourth most abundant element in the
universe. Atomically, carbon is a non-metallic element which, in
addition to the amorphous form, occurs in several
crystalline structures: graphite (solid
- hexagonal structure), diamond
(cubic structure, forming very hard, optically transparent
crystals), fullerene (spherical "molecules"
from networks of carbon atoms, e.g. C60).
Carbon is extremely important in
terrestrial nature (content in the earth's
crust approx. 200-800 mg/kg), where it
occurs mainly in compounds, the most important of which is
gaseous carbon dioxide CO2
(approximately 0.04% in the Earth's atmosphere - it is important
for photosynthesis in plants), and of
minerals, as calcium carbonate CaCO3. CO2 and some other carbon gases (e.g.
methane) show a considerably high
reflective capacity for thermal infrared radiation ("greenhouse effect")
and thus act as a thermoregulatory component in the atmosphere of
the Earth and other planets.
Thanks to the specific configuration of
the atomic shell, carbon is able to form a huge amount of
so-called "organic" compounds
with hydrogen, oxygen, nitrogen, phosphorus and other elements,
often to form very long, branched and cyclic molecules. Thanks to
these very diverse combinations, which can react together again
to form other complex specific substances, carbon compounds have
become the basic building block of living matter - carbon is an
essential biogenic element.
Carbon has a number of isotopes (8C
- 22C), of which only two isotopes 12C (98.93%) and 13C (1.07%) are stable. Important are two
radioisotopes of carbon :
Carbon 14 C
From the radioactive carbon isotope is most important radiocarbon
14C6, which with a half-life of 5730 years
converts by b- -radioactivity 14C ® 14N + e- + n' to the ground
state of nitrogen 14N7.
14C is a pure
beta emitter with a maximum energy of emitted electrons
156.5 keV (blue curve in the right part of
Fig.1.4.7 mentioned above in passage 3 H). It occurs in nature as an important cosmogenic
radionuclide, which is formed by the action of neutrons
ejected by cosmic radiation from the nuclei of atoms, on nitrogen
in the upper layers of the Earth's atmosphere: no + 14N7 ® 14C6
+ p+ (see §1.6, section "Cosmic radiation", Fig.1.6.7). It is the
basis of the radicarbon method of determining
the age of archaeological objects
(described above in the section "Radioisotope (radiometric) dating", passage "Radiocarbon dating method").
The content of radiocarbon-14 in nature is also
influenced by human activity. To a lesser extent there is a reduction
in the content of 14C in the environment by burning fossil fuels,
in which over millions of years, the original content 14C has already
disappeared by radioactive decay; thus, CO2 generated by the combustion of fossil fuels does not
contain 14C
and the proportion of natural radioactive 14CO2 is thus diluted. Nuclear technologies, on the
other hand, increase the content of radiocarbon-14. The 14C content increased
significantly in the 1960s during nuclear weapons tests due to
the 14N
(n, p) 14C
nuclear reactions by the effect of neutrons released during the
explosion, on atmospheric nitrogen. A smaller amount of
radio-carbon 14C is also formed during the operation of fission
nuclear reactors by the nuclear reaction 17O (n, a) 14C on oxygen in
cooling water and by reacting 14N (n, p) 14C on nitrogen dissolved in cooling water; from the
cooling water with ventilation, this small amount of 14CO2 gets into the air
through the ventilation chimneys of the power plant.
Like tritium, 14C radio-carbon is
produced artificially by neutron activation of 14N (n, p) 14C in a nuclear
reactor for many applications, mainly biological ones, especially
tracking methods (§3.5 "Radioisotope
tracking methods").
Carbon 11 C
Another somewhat important radioisotope of carbon is short-lived positron
11C, which with a half-life of
20.36 min. converts mainly by beta+
-radioactivity (99.75%) and to a small extent by
electron capture (0.25%) to the ground state of boron 11B. It is used
(relatively rarely) in positron emission tomography
(§4.3 "PET cameras", § 4.8 "Radionuclides
and radiopharmaceuticals for scintigraphy"). The isotope 11C is prepared in a
cyclotron either by irradiating boron with accelerated deuterons
in reactions 10B (d, n) 11C, 11B (d, 2n) 11C (taking place simultaneously
with both stable isotopes of natural boron),
or by irradiating nitrogen with protons: 14N (p, a) 11C.
In this connection
we will mention three other light positron radionuclides
used in positron emission tomography :
Nitrogen,
Oxygen, Fluorine
Nitrogen N 7
( Nitrogenium ) is an inert
gaseous element at normal temperatures, forming the main part of
the Earth's atmosphere (78% by volume). At -195 °C it changes to
a liquid state; liquid nitrogen is a very important cryogenic
medium. Along with carbon, hydrogen and oxygen, nitrogen is an
important biogenic element as part of the amino
acids that make up proteins. Nitrogen has two
stable isotopes 14N (99.64%) and 15N (0.36%). Radioactive isotopes of nitrogen have
relatively short half-lives, less than 10 minutes. It is worth
mentioning two of them :
Nitrogen 13 N
with a half-life T1/2
= 9.96 min. converts
by beta+ -radioactivity (99.82%) and to a small
extent by electron capture (0.18%) to the basic state of carbon 13C. The 13N isotope is prepared
in a cyclotron either by irradiating oxygen (in water) with
accelerated protons by a nuclear reaction of 16O (p, a) 13N, or by irradiating
carbon with deuterons in the reaction 12C (d, n) 13N. Nitrogen 13N is occasionally used in positron emission
tomography in the form of ammonia and 13N-labeled amino acids.
Note: Isotope 13N played an important
role in the process of studying radioactivity: it was the first
artificially prepared radioactive isotope. In 1934, F.Joliot and
I.Curie discovered it in experiments with irradiation of boron
with alpha particles (from polonium); was formed here by the
reaction of 10B (a , n) 13N.
Nitrogen 16 N
with a half-life T1/2
= 7.13 sec. converts
beta-- radioactivity (Ebmax = 4290keV and 10420keV) to the ground state (28%) and
to the highly excited states of 6130keV (67%) and 7115keV (5%) of
oxygen 16O.
During deexcitation, high-energy gamma photons 6.13MeV and 7.1MeV
are emitted. Isotope 16N is formed by the reaction of 16O (n, p) 16N fast neutrons with the basic oxygen isotope 16O, mainly in the
cooling water of the primary circuit of water-cooled nuclear
reactors. The activity of the 16N isotope in the circulating cooling water is
proportional to the number of neutrons in the reactor core, i.e.
the intensity of the chain fission reaction (the number of
fissiones per second) and the thermal power of the reactor.
Measurement of 16N activity by means of gamma-detectors attached from the
outside of the cooling water circulation pipe of the primary
circuit can therefore monitor the correct operation of the
reactor.
Oxygen O 8
(Oxygenium) is a reactive gaseous
element at normal temperatures, making up 21% (by volume) of the
Earth's atmosphere. Together with carbon and hydrogen, it is the
most important biogenic element. It has three
stable isotopes: 16O (99.76%), 17O (0.04%), 18O (0.2%). The radioactive isotope 15O is sometimes used in positron emission tomography :
Oxygen 15 O
with a half-life T1/2
= 122 sec. converts
by beta+ -radioactivity (99.82%) and to a small
extent by electron capture (0.12%) to the basic state of nitrogen
15N.
Isotope 15O
is prepared in a cyclotron either by irradiating nitrogen
(enriched with the 15N isotope ) by accelerated deuterons in a nuclear
reaction of 15N (d, 2n) 15O, or by irradiating oxygen with protons in a 16O (p, pn)15O reaction. Its use
in PET is relatively rare due to the short half-life.
Fluorine
F 9 is a green-yellow gas at normal temperatures, very
reactive, so it occurs on Earth only in compounds. It belongs to
the group of halogens - salt-forming elements (Greek halos = salt). It
is relatively widespread (540mg / kg in the
earth's crust). It has a single stable
isotope of 19F. Radioactive isotope 18F is very widely used in positron
emission tomography :
Fluorine
18 F
with half-life T1/2
= 110 min. converts
by beta+ -radioactivity (96.86%) and electron
capture (3.14%) to the basic state of oxygen 18O. The isotope 18F is prepared in a
cyclotron most often by irradiating of oxygen (in water enriched with the isotope 18O) by accelerated
protons in reaction 18O (p, n) 18F, less often by irradiation of compressed neon with
accelerated deuterons by nuclear reaction 20Ne (d, a) 18F. In positron emission tomography PET
is mostly used in the form of 18F-fluoro-deoxyglucose (FDG - §4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy",
part "Positron radionuclides").
Positron radionuclides
All positron radionuclides, if they are contained in the
substance environment (which is practically
always), emit gamma radiation with an
energy of 511 keV (§1.2,
part "Radioactivity b+ ") during the annihilation
of emitted positrons with electrons e+ + e- ® 2 g (§1.2, part "Radioaktivity b+" and §1.6, section "Interaction of charged particles- direct ionizing radiation", Fig.1.6.1 below part). The
gamma-spectrum of "pure" light positron radionuclides,
which we have discussed so far, is formed by a single annihilation
e+e- peak 511keV *) with an intensity close
to 200% - in 11C (199.5%), 13N (199.6%), 15O (199.7%), 18F (193.7%). Some other
(heavier) positron radionuclides have a positron content of less
than 100% and therefore the intensity of annihilation radiation
is often significantly lower than 200% - see eg 22 Na (180.7%), 58Co (29.8%), 64Cu (35%), 68 Ga (177.8%), 89 Zr (45.6%); other gamma peaks
are also present, from nuclear deexcitations.
*) It is interesting that the annihilation gamma peak 511keV is
slightly wider in the spectrum, compared to the
surrounding "nuclear" photopeaks, due to the Doppler
expansion caused by different braking rates of positrons in
the substance before annihilation with electrons (discussed in §1.2, passage "Spectrum gamma radiation"). This extension is only
about 1.2 keV and is manifested only on semiconductor spectra (we have clearly observed them in our detailed
spectrometric measurements, but these differences are not visible
on the strongly grapfically reduced and contracted spectra
displayed below...).
Conversion schemes of positron radionuclides 11C, 13N,
15O, 18F and gamma spectrum of their
annihilation radiation 511 keV.
Positron radionuclides are used mainly in nuclear
medicine in the form of suitable radiopharmaceuticals,
which are captured and accumulated especially in tumor tissues,
the distribution of which is then imaged by PET positron
emission tomography based on the coincidence
detection of gamma 511keV annihilation photon pairs (see §4.3 "Tomographic
cameras" and §4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy",
part "Positron radionuclides"). Of the above-mentioned
light positron radionuclides, only fluorine 18F
is widely used, other positron radionuclides (C-11, N-13, O-15)
only rarely, due to their short half-life.
In our treatise we will gradually present a number of
other important light, medium and finally heavy nuclides :
Phosphorus,
Sulfur
These are important biogenic elements, abundant in the earth's
crust, seawater and living organisms.
Phosphorus P 15 (lat. Phosphorus) is a relatively widespread
non-metallic element - content in the earth's crust about 1 g/kg.
Pure phosphorus occurs in several allotropic modifications, the
most famous of which are three named according to their color
appearance: White (yellowish) phosphorus formed by P4
molecules, which is soft and highly reactive, especially with
oxygen (can spontaneously ignite). Red phosphorus with a polymer
structure that is relatively stable. Black phosphorus with a
layered polymer structure is the most stable and also exhibits
metallic electrical, thermal and optical properties. In nature,
however, phosphorus is only found in compounds.
Phosphorus is a very important biogenic element found in all plant and animal cells. It
is a binding phosphate component of genetic biomolecules DNA and
RNA, energy molecules adenosine triphosphate, phospholipid cell
membranes (§5.2,
part "Cells - basic units of living
organisms"). It is also stored in bones in calcium
phosphate, hydroxiapatite.
Phosphorus has a single stable isotope 31P. From a number of radioactive isotopes 24-46P most have short
half-lives (minutes, seconds, milliseconds)
. Due to the longer half-life, only two
phosphorus radioisotopes are important :
Phosphorus 32 P
With a half-life of 14.27
days, is converted by beta- radioactivity (Eb max = 1710 keV) to sulfur 32S in the ground state, it is a pure beta emitter.
In nature, a small amount of the 32P isotope is formed as a cosmogenic radionuclide in the
atmosphere by nuclear reactions of secondary neutrons from cosmic
rays. It is produced artificially in a nuclear reactor by
irradiating of phosphorus in the reaction 31P (n, g) 32P, or by irradiating sulfur-32 with fast neutrons by the
reaction 32S
(n, p) 32P.
Radioactive 32P is used as a radioindicator in
laboratory methods of molecular biology and genetics for
radiolabeling. 32P -phosphate groups are incorporated into genetically
important molecules of nucleotides, DNA, RNA, which can then be
sequenced and detected radiometrically (autoradiographically) -
§2.2, passage "Autoradiography". 32P can also be used to diagnose and treat cancer - tumor
cells tend to take up more phosphorus compounds than normal
cells.
Radiophosphorus 32P, as a pure beta-radionuclide, was used for the
treatment of hematological malignancies (iv application of about 200-500 MBq in the form of
phosphate), especially polycythemia
- §3.6 "Radioisotope therapy", passage "Treatment of hematological
diseases of radiophosphorus 32P".
Phosphorus
33 P
is converted with a half-life of 32.97 days by beta- radioactivity (Eb max = 248 keV) to sulfur 33S in the ground state. Radioactive 33P is used in similar laboratory applications as
phosphorus-32, in methods where lower beta electron energy is
preferred (eg better resolution in autoradiography).
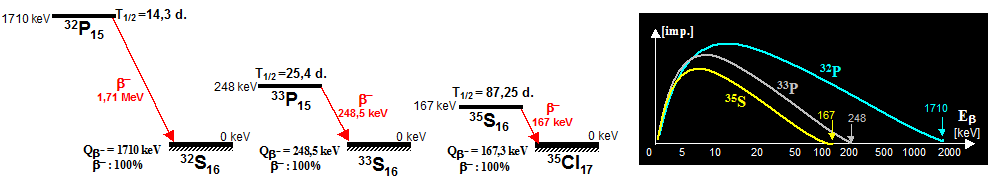
Conversion schemes of radionuclides 32P,
33P, 35S
and their beta spectra (the same note applies to
the beta spectrum as in Fig.1.4.7) .
Sulfur S 16 (lat. Sulfur) is also
highly reactive and therefore occurs in nature above all in
compounds, mainly in sulfides. Contents in the earth's crust of about
0.03 to 0.09 %, concentration in sea water of about 900
mg/liter. As a pure element, it occurs during volcanic activity,
where it crystallizes from hot volcanic gases containing sulfur
fumes. Melting point 115°C, boiling point 445°C. Sulfur is
also an important biogenic
element, it is part of amino acids (cysteine, methionine) forming
proteins.
It has four stable isotopes: 32S (94.99%), 33S (0.75%), 34S (4.25%) and 36S (0.019%). Of the
radioactive isotopes of sulfur, one has a practical application :
Sulfur
35 S
is converted with a half-life of 87.25 days by beta- radioactivity (Eb max = 167.3 keV) to chlorine 35Cl in the ground state. Like 32P, 35S sulfur is formed in small amounts in the atmosphere as
a cosmogenic radionuclide. The 35S isotope is artificially prepared in a nuclear reactor
by irradiating chlorine (potassium
chloride) with neutrons by reacting 35Cl (n, p) 35S. Radioactive sulfur-labeled amino acids 35S-cysteine and 35S-methionine is used
in biochemical research to study amino acid metabolism.
Sodium,
Potassium
Sodium and potassium are the most common elements of the alkali
metals, abundant in the earth's crust, seawater and
living organisms. They are soft (they can be
cut with a knife),
light (they float on water), silvery shiny metals, easily
melted (sodium has a melting point of
97.7°C, potassium 63.4°C). They are very
reactive (they react particularly violently with water), so they occur in nature only in
compounds.
Sodium Na 11 (lat. Natrium) is the most common alkaline
element - content in the earth's crust 2.4-2.6 %,
concentration in seawater 10.5 g/liter. Sodium is one of the biogenic elements and is found in all cells of plant and
animal tissues, as well as in extracellular fluid, cca 4% of salt NaCl.
It has a single stable isotope of 23Na. Of the
radioactive isotopes, sodium-22 is particularly important :
Sodium
22 Na
is converted with a half-life of 2.6 years
predominantly by beta+
-radioactivity (90%, Eb max = 500keV) and partly by electron capture
(10%) to 22Ne
in the excited state of 1275keV; during deexcitation, gamma
photons with the same energy of 1275keV are emitted. Only 0.056%
are converted to baseline 22Ne. The isotope 22Na is prepared in a cyclotron either by irradiating
magnesium metal with deuterons in the nuclear reaction 24Mg (d, a) 22Na, or by irradiating
fluorine with alpha-particles in the reaction 19F (a,n) 22Na.
The annihilation peak 511 keV
(180%) dominates in the gamma spectrum of sodium-22, in the area
of higher energies there is a significant peak 1274 keV (100%).
Conversion scheme and gamma spectrum of sodium 22Na.
Radionuclide 22 Na is mainly used as a laboratory source of
positrons for atomic and nuclear physics (see eg §1.5, section "Antiparticles - antiatoms - antimatter -
antiworlds", passage
"Artificial production of antimatter")
and for material analysis of samples *).
For some PET cameras, sealed Na-22 emitters serve as calibration
and adjustment sources of 511 keV annihilation gamma radiation (in addition to 18F samples and Ge 68
-Ga 68 standards ) - see
§4.3, section "Positron emission tomography
of PET".
*) Analysis of materials using positrons,
so-called positron annihilation spectrometry (briefly described in §3.3, section "Positron annihilation spectrometry"), is based on the behavior
of positrons as they pass through the test substance. The lifetime
of the positron is measured, ie the time interval between
its formation in beta+ -radioactivity of 22Na (indicated by photon emission
1275keV) and extinction (accompanied by radiation of annihilation photons
511keV), which can provide information
about structural irregularities - dislocations, vacancies,
impurities in crystal structure.
The alkali metal potassium K 19 (lat. Kalium) is also
very reactive and therefore occurs in nature only in compounds. Contents in the crust of about
2.0 to 2.4 %, concentration in sea water of 380
mg/liter. Potassium is also a biogenic
element.......
It has two stable isotopes 39K (93.3%) and 41K (6.73%). Natural
potassium also contains 0.0017 % of the long-lived radioactive
isotope 40K :
Potassium
40 K
is a widespread natural (primary) radionuclide
with a very long half-life of 1,25.10 9
years. Decays branched by beta- -radioactivity (89%) to the
ground state of calcium 40Ca and by electron capture
(11%) to argon 40Ar - to its excited state of 1460 keV. Both of these
daughter isotopes are stable, further decay no longer continues.
The characteristic gamma peak of 1460 keV
(10.5%) of potassium is clearly visible in all spectrometric
measurements of natural samples with an acquisition time of
several hours (cf. Fig.1.4.2 above in the section "Natural radionuclides").
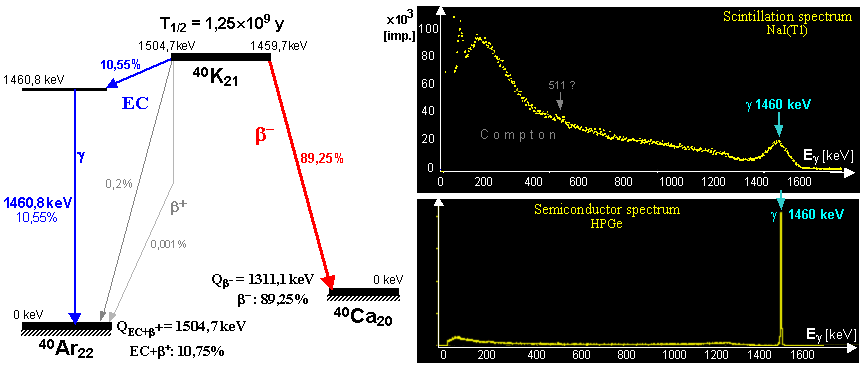
Conversion scheme and gamma spectrum of
potassium 40K.
Scandium
Scandium Sc 21
(the name comes from its discovery in Scandinavia - L.F.Nilson,
1879, Sweden) is a
silvery white soft and light metal somewhat similar to aluminum,
its proportion in the earth's crust is about 5-2 mg/kg. It has a relatively small use, it is used in alloys
with aluminum. It
has a single stable isotope of 45Sc. Of the about 25 known radioactive
isotopes of scandium, 44Sc and 47Sc have
promising applications in nuclear medicine :
Scandium
44 Sc
Transforms with a half-life of 3.97
hours, predominantly beta+ -radioactivity (94%, Eb+ max = 1474keV) and partially electron capture
(5.7%) to 44Ca in the excited state of 1157keV (higher
excited levels of 2656 and 3301 keV are only weakly represented); during deexcitation, gamma photons are emitted mainly
with this energy 1157keV. The annihilation peak 511keV
(188%) dominates in the gamma spectrum of scandia-44, in the area
of higher energies is the peak 1157keV (99.8%); faint higher g -peaks 1499, 2144, 2556 and 3301 keV are only visible at
high magnification.
Conversion scheme and gamma spectrum of scandium 44Sc.
Positron radionuclide 44Sc has promising use
in nuclear medicine for scintigraphic imaging by PET
positron emission tomography (see
§4.3 "Tomographic cameras" and §4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy",
section "Positron radionuclides"). Trivalent skanium binds well, using proven
bifunctional chelating agents of the DOTA type, to
vector biomolecules, mainly monoclonal antibodies. Compared to 18F and especially 68Ga, a longer
half-life of almost 4 hours is also advantageous, allowing
imaging even with heavier biomolecules with slower
pharmacokinetics. It can also be used theranostically
in combination with analogous therapeutic
radiopharmaceuticals labeled with 90Y, 177Lu, or ideally 47Sc.
Its great advantage is the production
method: Although it can be produced by nuclear reaction of 44Ca (p, n) 44Sc on a small
cyclotron, in nuclear medicine workplaces (without
the need for a cyclotron) 44Sc
can be continuously and long-term obtained by elution from 44Ti/44Sc
generator :
The parent titanium 44Ti for the generator is obtained on a
cyclotron by a nuclear reaction of 45Sc (p, 2n) 44Ti. In the column of the generator, the parent 44Ti with a long half
- life of 60.4 years is transformed by electron
capture into the daughter 44Sc, which is eluted with a suitable solution (H2C2O4 + HCl). Due to the long half-life of the parent 44Ti (approximately 130,000 times longer than the daughter 44Sc), secular equilibrium is maintained in the
generator (see section "Radionuclide
Generators"), so that in the long run, for many years, the required 44Sc can be eluted
every day, with the same activity, given by the activity of the
default 44Ti...
Given these
advantageous properties, it can be expected that scandium 44Sc
can in come cases replace the existing major PET
radionuclides 18F and 68Ga ..?..
A certain disadvantage of 44Sc is the
simultaneous emission of high-energy gamma radiation 1157keV,
which increases the radiation dose of patients and PET workers.
Therefore, there are considerations about the use of the
neighboring radionuclide scandium 43Sc (T1/2 3.89 hours, beta+ 88%, gamma 373keV (23%)), which has properties similar
to 44Sc,
but shows much lower energy and intensity of the accompanying
gamma radiation ...
Scandium
47 Sc
With a half-life of 3.35 days, it is converted
by beta- -radioactivity to 47Ti (which is a stable nuclide), in
31.6% to baseline and in 68.4% to an excited level of 159 kV.
Max. the energy of the beta electrons for both cases is 600keV
and 441keV, the mean beta energy is 162keV. The 47Sc gamma spectrum
consists of a single 159keV peak (68%).
Conversion scheme and gamma spectrum of scandium 47Sc.
The 47Sc isotope can be prepared by neutron reactions 47Ti (n, p) 47Sc, 46Ca (n, p) 47Ca .. ... 47 Sc .... ... in a
nuclear reactor, or in a cyclotron by reactions 48Ca (p, 2n) 47Sc, 46Ca (p, g) 47Sc, ........
Scandium 47Sc is a promising beta- radionuclide for
therapy in nuclear medicine. In this case, the
gamma radiation of 159 keV can advantageously be used for scintigraphic
imaging (planar/SPECT) and monitoring of radionuclide
therapy. Sequence 44Sc/47Sc can be an excellent
teranostic isotope pair for PET imaging + radionuclide
therapy.
Chrome,
Iron
Chromium Cr 24 is a light white hard metal,
resistant to corrosion. It is relatively abundant in terrestrial
nature (in the earth's crust about 0.1-0.2
g/kg). It is mainly
used in metallurgy as an alloying additive or for plating. It has
4 stable isotopes: 50Cr (4.35%), 52Cr (85.8%), 53Cr (9.5%), 54Cr (2.37%) .
Of the radioactive isotopes, one found practical application :
Chromium
51 Cr
It is converted with a half-life of 27.7 days by electron
capture to the stable isotope 51V - in 90% of the transformations it is to the ground
state and in 10% to the excited state 320keV, during the
deexcitation of which gamma photons of the same energy are
emitted. The very simple gamma spectrum of chromium-51 consists
of a single 320keV photopeak (10%). Compared to
most other beta (EC) -gamma radionuclides, 51-chromium is, in
recalculation on the activity value, a relatively weak
gamma emitter, about 10 times weaker than cesium 137Cs, cobalt 60Co or radioiodine 131I. Isotope 51Cr is prepared by
nuclear reaction 50Cr (n, g)51Cr by irradiating chromium (enriched
to about 80% with the isotope 50 Cr) by neutrons in a nuclear reactor.
Decomposition scheme and gamma spectrum of chromium 51Cr.
The radionuclide 51Cr is used as a tracer radioindicator
in some applications in industry, geology and nuclear medicine.
In nuclear hematology, by means 51Cr-chroman ions were labeled the red blood cells,
or event. white blood cells. Using the autologous erythrocytes
labeled in this way, the lifespan (or half-life) of the
blood cells and, if appropriate, the place of their
destruction (§4.9 "Survival and
sequestration of erythrocytes"). The use of 51Cr in nuclear medicine is almost abandoned.
Radionuclide 51Cr
was too used several times to track groundwater pathways. A
solution of radiochrome was poured into an immersion spring,
after which water samples were taken from several nearby
watercourses and measured on a sensitive well scintillation
detector - whether 51Cr radioactivity appeared in
them, for what time and in what quantity.
Iron
Fe 26 (Ferrum) is a ferromagnetic
metal element, relatively abundant in nature (4-6% in the earth's
crust). This well-known metal is a basic construction material in
mechanical engineering, construction, electrical engineering and
many other technical fields, as well as in household and everyday
life. It has four stable isotopes: 54Fe (5.8%), 56Fe (91.7%), 57Fe (2.2%) and 58Fe (0.28%). Of the many radioactive isotopes of iron,
two are of practical importance :
Iron 55 Fe
is converted with a half-life of 2.74 years by electron
capture to the ground state of manganese 55Mn. The
characteristic X-rays Ka,b with energies
around 6keV are emitted. 55Fe
emitters with sufficiently high activity are used as sources of
soft X-rays for X-ray fluorescence analysis of
light elements (lighter than manganese), eg in geological survey
of rocks. Iron 59 Fe
It is converted with a half-life of 44.5 days of beta- -radioactivity to stable cobalt 59Co, mainly to excited
states of 1099keV (53%) and 1292keV (45%). During deexcitation, g radiation is
emitted mainly with these two energies (with
low representation then 142, 192, 335, 1482 keV). It was used in ferrokinetic tests in hematology
in nuclear medicine to study iron metabolism in connection with
the processes of red blood cell production in anemia or leukemia (§ 4.9.9. , passage "Examination
of iron kinetics with 59 Fe"). In plant biology, the isotope 59Fe has been used to monitor the uptake of iron from soil
and nutrient solutions and its storage in various parts of
plants.
Cobalt
Cobalt Co 27 is a ferromagnetic metal element
relatively similar to iron, its proportion in the earth's crust
is about 25 mg/kg. Its main use is in
metallurgy. It has
only one stable isotope 59Co. The most important cobalt radioisotopes
are 60Co
and 57Co :
Cobalt
60Co
Of the medium-heavy radionuclides, cobalt 60Co
in particular is widely used as a source of hard
radiation with gamma energies of 1173 + 1332 keV
for radiotherapy (§3.6,
part "Isocentric
radiotherapy"), defectoscopy
(§3.3, part "Radiation defectoscopy") and other technical
application. 60Co, with a half-life of 5.27
years, is transformed by beta- -radioactivity into excited states of
nickel 60Ni
(which is a stable nuclide). It is mainly, in 99.88%, the level
of 2506 keV, which cascades deexcites first to the level of
1332.5 keV, which then to the ground state of 60Ni. The spectrum of
gamma radiation is formed by two peaks 1173 and 1332 keV, in the
scintillation spectrum the continuous Compton scattered radiation
is also significantly shown.
The 60Co isotope is prepared by neutron irradiation of cobalt
metal in a nuclear reactor by the reaction of 59Co (n, g) 60Co.
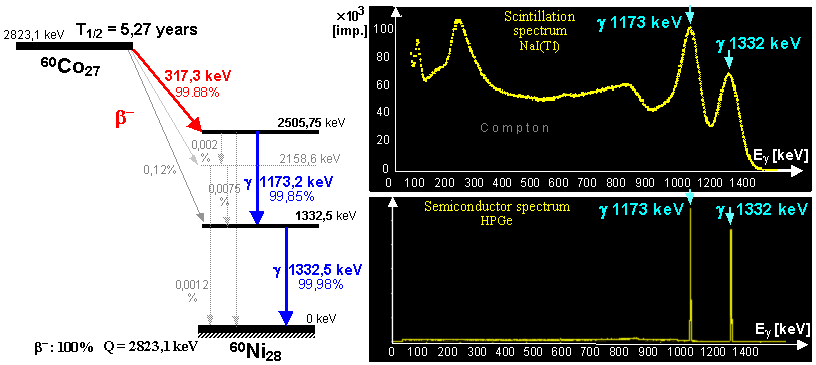
Conversion scheme and gamma spectrum of
cobalt 60Co.
Cobalt
57 Co
Another important radioisotope of cobalt is 57Co, which is used as a source of softer
gamma radiation 122 +136 keV. 57Co with a half-life
of 271.8 days is converted by electron
capture to excited levels of 706.4keV (0.18%) and
especially 136.5keV (99.82%) of iron 57Fe. The most saturated level of 136.5 keV is deexcited
in 10% to the ground state of 57Fe and in 85.5% to the level of 14.4keV with the
emission of main energy gamma 122 keV. After
electron capture, photons of the characteristic X-ray Ka,b are
emitted during electron jumps between the L and K
shells of iron with energies of 6.4-7.1 keV, as well as
conversion and Auger electrons. Gamma spectrum 57Co consists above all
of the main peak 122keV (85.5%), with the lower neighboring peak
136keV (11%); in the area of higher
energies, a very weak peak of 692keV (0.16%) can be seen only
after high magnification.
Isotope 57Co is prepared by deuterone irradiation of metallic iron
in a cyclotron by a nuclear reaction of 56Fe (d, n) 57Co.
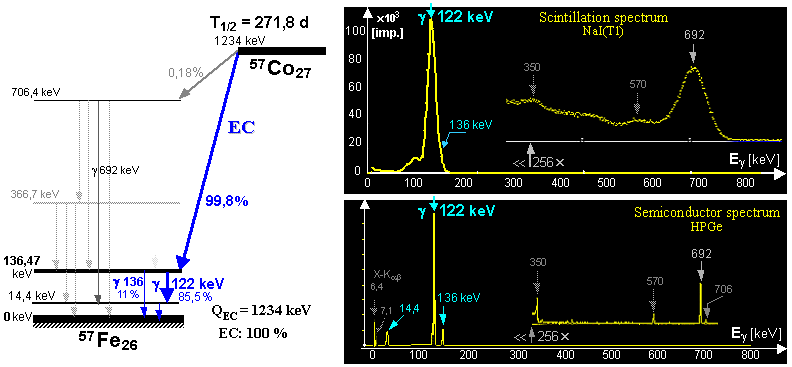
Conversion scheme and gamma spectrum of cobalt 57Co.
57Co is used
as a source of gamma radiation for Mösbauer
spectrometry of iron and its compounds (§3.4,
part "Mössbauer
spectroscopy"). In nuclear medicine, vitamin B12
labeled 57Co was used to determine the intake of this vitamin by
intestinal resorption (§ 4.9.9, passage "Schilling
test").
Both mentioned cobalt radionuclides 57,60Co
are also used as reference gamma emitters. For
the proximity of gamma radiation energy 122keV with energy 140keV
99mTc,
with point sources 57Co they are used as pointers in
scintigraphy and planar homogeneous sources 57Co are used to adjust
and test the homogeneity of the field of view of scintillation
gamma cameras (see "Phantoms
and phantom measurements in nuclear medicine").
Cobalt 58Co
For radioisotope diagnosis in nuclear medicine in the 60s-80s was
also used the cobalt 58Co, which is a 70.8 day half-life of
the electron capture (84%), and b+
-radioactivity (15%) is converted to the excited states
of iron 58Fe - 811keV (84 + 15%) and 1675keV (1.2%).
During deexcitation, gamma photons are emitted mainly with an
energy of 811keV (99.4%), with a much lower intensity of 864keV
(0.7%) and 1675keV (0.5%). In nuclear physics, it is sometimes
used as a source of positrons. In nuclear medicine,
vitamin B12 labeled 58Co
was used in the 1970s and 1980s, similar to the above 57Co.
All these methods are now abandoned.
Nickel 56Ni
--> Cobalt 56Co
In a somewhat atypical way, we present here the following two
interrelated radionuclides, which are important in nuclear
astrophysics during supernova explosions :
Nickel 56Ni
according to astrophysical analyses, it is produced in colossal
quantities (on the order of the mass of our
globe..!..) during the explosion of
supernovae, especially thermonuclear supernovae
of type Ia - it is described in §4.2,
section "Supernova
explosion. Neutron star. Pulsars." monograph "Gravity, black holes and
space-time physics".
With a half-life of 6.1 days,
56Ni is converted by electron
capture to the excited 1.72 MeV level of cobalt 56Co. During
deexcitation through the three lower levels, gamma radiation with
energies of 158keV (99%), 269keV (37%), 480keV (36%), 750keV
(49%), 812keV (86%), 1562keV (15%) are emitted.
The daughter 56Co is also radioactive :
Cobalt 56Co
with a half-life of 77.27 days is converted by beta+-radioactivity (20%, Eb+max=1.49 MeV) and electron
capture (80%) to relatively high excited states of iron 56Fe
(2085keV (20%), 3120keV (15%) , 3451keV
(21%), 3830keV (15%), 4050keV (24%), 4300keV (1.4%)), which is stable. During deexcitation, hard gamma rays
of many energies are emitted, the most prominent of which are
847keV (100%), 977keV (1.4%), 1038keV (14%), 1175keV (2.3%),
1238keV (67%), 1360keV (4.3%), 1771keV (15%), 2015keV (3.1%),
2035keV (8%), 2598keV (17%), 3009keV (1%), 3202keV (3.2% ),
3253keV (8%), 3451keV (1%).
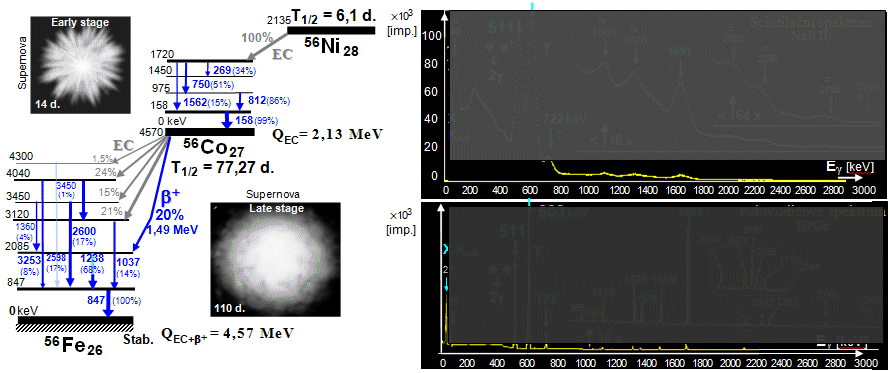 .
.
Transformation scheme and gamma spectrum of 56Ni and 56Co (the spectrum will be measured
and refine the decay scheme, when we get a sample of 56Ni...).
Both of these radionuclides 56Ni and 56Co emit a huge amount
of energy by their radioactive decay in supernovae, thanks to
which the supernova can shine for many days with the brightness
of billions of Suns (passage "Supernova radiation. Light curve. Spectrum of
radiation")
..!..
These radionuclides are of no importance
for technical or laboratory applications.
Copper,
Galium, Germanium, Selenium
Copper Cu29 (Cuprum) is
a well-known yellow-red shiny metal with very good electrical and
thermal conductivity. The content in the earth's crust is about
50-70 mg/kg, pure metallic copper occurs only
rarely, it is mostly found in compounds - sulfides, oxides, in
combination with iron compounds and others. In addition to
metallurgy (alloys with tin - bronze or
zinc - brass),
mechanical engineering and construction, the main use of copper
in electrical engineering
is due to excellent
electrical conductivity and corrosion resistance. Copper has two
stable isotopes, 63Cu (69.15%) and 65Cu (30.85%). Some radioactive isotopes
of copper are sporadically used or tested in nuclear medicine
(especially PET scintigraphy) and biochemical studies: 61Cu (T1/2 = 3.3 hours, b+, EC), 62Cu (T1/2 =
9, 7min., b+, EC), 64Cu (T1/2 = 12.7 h, b+, EC), 67Cu (T1/2 =
61.8 h, b-, g). Two copper
radionuclides appear promising :
Copper
64 Cu,
which with a half-life of 12.7 hours is converted by beta+ -radioactivity (17.5%) and electron
capture (44%) to the basic (53%) and excited (0.5%)
state of 64Ni;
further beta- -radioactivity (38.5%) to the basic
state of 64Zn.
In the gamma spectrum of 64Cu dominates (as with any positron
radionuclide) e- e+ annihilation
peak 511 keV (35%), in the area of higher energies a
weak peak 1345.7 keV (0, 47%) can be seen only at a significant
magnification .
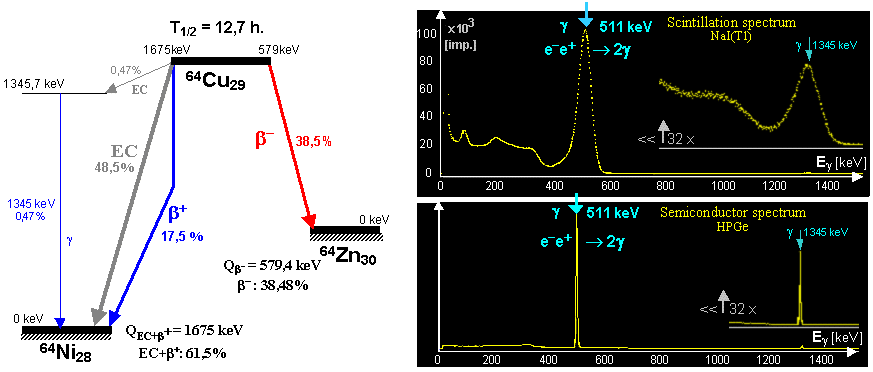
Conversion scheme and gamma spectrum of copper 64Cu .
The positron radionuclide Cu-64 is occasionally
used in positron emission tomography ("§4.3 "Positron emission
tomography PET",
§4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy",
section "Radionuclides and radiopharmaceuticals for
PET"). Complex 64Cu-ATSM (acetyl methyl-thiosemicarbazone) are rapidly
and selectively taken up in hypoxic tissues, used for diagnosis
of hypoxia myocardium and brain, as well as tumor hypoxia. 64Cu-TETA-octreotide
permits PET imaging neuroendocrine tumors. It is also tested for
labeling of monoclonal antibodies, e.g. 64Cu-DOTA-cetuximab, bombesin peptide.
Copper
67 Cu
is converted with a half-life of 61.8 hours by beta- -radioactivity to basic (20%) and
excited states of zinc 64Zn, during their deexcitation gamma radiation with
energies of 91keV (7%), 93keV (16%), 185keV (49%) is emitted;
three other higher energies up to 393keV are weak, <1%....
....... picture - decay scheme + gamma
spectrum ....add......
Combination of the designation of the same biochemical carrier
with the isotope Cu-64 for the diagnosis of PET
and Cu-67 for beta-rays therapy
are promising for theranostics in nuclear
medicine...
Galium Ga 36 is a soft, very easily fusible
metal (melts already at 30 °C) in a light blue-gray color. It occurs
relatively rarely (representation in the
earth's crust about 15 mg/kg). Its main use is in electronics as a
component of a number of semiconductor materials in the
manufacture of transistors and light emitting diodes (LEDs). It
has two stable isotopes 69Ga (60.1%) and 71Ga (39.9%). Two of the radioactive
isotopes are used :
Galium 67 Ga
With a half-life of 3.26 days, is transformed by electron
capture into excited states of 67Zn with energies mainly 93keV (52.5%), 184keV (22.7%)
and 393keV (23.6%), the basic state is only 3%. During
deexcitation of the daughter 67Zn, photons of gamma radiation with energies mainly of
93, 184 and 393 keV are emitted. The gamma radionuclide radiation
spectrum of 67Ga consists mainly of three main lines 93keV (38%),
184kev (21%) and 300keV (17%), the weaker line is 394keV (5%). In the higher energy region, weak peaks of 494, 794 and
887 keV (representation around 0.1%) are visible in the spectrum
only at high magnification.
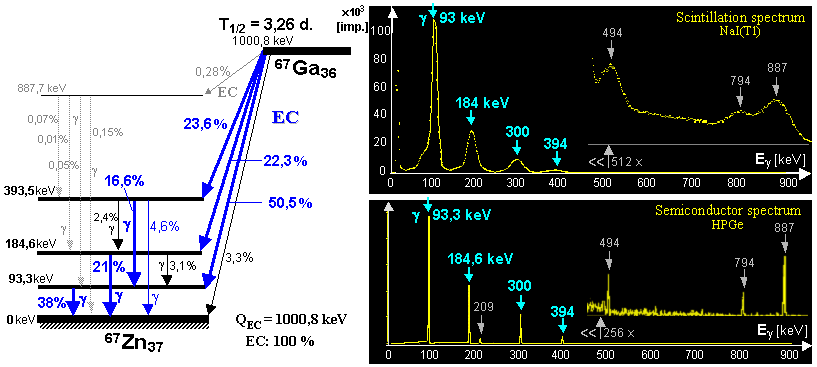
Decomposition scheme and gamma spectrum of gallium 67Ga.
The 67Ga isotope is prepared by irradiating zinc or copper in
a cyclotron with accelerated protons, deuterons or helium nuclei (a -particles) by nuclear reactions: 67Zn(p,n)67Ga, 68Zn(p,2n)67Ga, 67Zn(d,2n)67Ga, 65Cu(a,2n)67Ga.
67Ga is used in nuclear medicine (in the form of a gallium-citrate complex) to image sites in tissues and organs affected by inflammatory
processes or tumor foci (§4.8 "Radionuclides and radiopharmaceuticals
for scintigraphy").
Galium
68 Ga
is converted with a half-life of 67.7 minutes by electron capture
(8.7%) and mainly by b+-radioactivity
(87.9%) to 68Zn, most often to the ground state (just
over 1% of the transformations are to excited states 67Zn, mainly to the
level of 1077keV). The gamma spectrum of
gallium-68 is dominated (as with any
positron radionuclide) by e-e+ annihilation
peak 511 keV (178%), in the area of higher energies weak
peaks 1077 (3%), 1261
(0.1% ) and 1883 (0.14%) keV are visible only at
high magnification.
Conversion scheme and gamma-spectrum of gallium 68Ga.
Radiopharmaceuticals labeled with 68Ga are used in
nuclear medicine for PET imaging (§4.3 "Positron emission
tomography of PET",
§4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy",
passage "Radionuclides and radiopharmaceuticals for
PET"). The 68Ga isotope is used in workplaces most often obtained
from germanium-gallium generators 68Ge/68Ga
(mentioned a bit below in the passage
"Germanium 68
Ge", including a
germanium phantom for PET cameras). Galium 68 Ga is to some extent "PET analog" of
usual technetium 99mTc for SPECT and planar scintigraphy. Allows chelating
binding to a variety of radiopharmaceuticals, including lighter
monoclonal antibodies (of Fab fragments).
So far, the most frequently used for display of neuroendocrine
tumors (somatostatin receptors -
labeled octreotide type 68Ga-DOTATOC and its derivatives).
Very promising seems PET imaging of prostate tumors using labeled
prostate membrane antigen 68Ga-PSMA (J591,
617, I & T, ...), followed by
biologically targeted radionuclide therapy using the same
antiPSMA monoclonal antibodies, labeled with beta or alpha
radionuclides.
Imaging of peptide
receptors expressing gastrin- bombesin receptors , eg
with 68Ga-DOTA-PEG2 -bombesin, 68Ga-BZH3 , is
also tested. Furthermore, epidermal growth factor receptors HER
using the labeled Fab
fragment of trastuzumab, also
vascular endothelial growth factor receptors VEGF. 68Ga-Annexin
can be used for PET to image apoptosis.
Galium-68 is a very promising diagnostic PET radionuclide, which
has - in combination with therapeutic radionuclides 90Y, 177Lu, 225Ac, 227Th - high teranostic
potential (discussed in §4.9, passage
"Combination of diagnostics and therapy -
teranostics").
Germanium Ge 36 is a gray-white solid semi-metallic
element, which occurs relatively rarely in nature (representation in the earth's crust approx. 5-7 mg/kg) . Its main use is in electronics as a component of a number of
semiconductor materials in the manufacture of transistors,
diodes, integrated circuits; Germanium
semiconductor detectors (Ge (Li), HPGe - §2.5 "Semiconductor detectors") are important for our field of nuclear
physics. Germanium has 4 stable isotopes: 70Ge (20.4%),
72Ge (27.3%), 73Ge (7.8%)
and 74Ge
(36.7%).
Natural germanium also contains 7.83% of the very long- lived radioactive isotope 76Ge (T1/2 = 1.7.1021 years; its radioactivity is negligible, it can be
considered almost stable - see below). Of
the radioactive isotopes, we mention three - one practically
used, the second used in an interesting experiment and the third
remarkable for its "exotic" way of decay :
Germanium
68 Ge
is converted with an half-life of 270.9 days by electron
capture to the ground state of the positron radionuclide
68Ga.
Based on this radioactive transformation is a 68Ge/68Ga
radionuclide generator, from which a short-lived
radioisotope gallium 68Ga (half-life of
68 minutes - difficult transport to places outside the cyclotron)
can be continuously obtained at nuclear
medicine workplaces for use in PET scintigraphy ( 68
Ga mentioned above). The Ge/Ga generator based on the principle of ion
chromatography is of a similar type as the known Mo/Tc
generator (described below - 99m Tc ). It is a glass chromatographic column in which the
parent 68Ge
is adsorbed in insoluble form on titanium oxide, aluminum or tin
oxide. They tested also "metal-free" carriers in
organic basis, such as trihydrohybenzene (pyrogallol)
formaldehyde resin, and porous sorbents based on nanomaterials
could be promised (zirconium and cerium
nanomaterials are being tested). After the
radioactive conversion of the 68Ge nuclei to the 68Ga daughter nuclei, the gallium atoms formed are
exported from the insoluble bond and can be eluted
from the column by weak hydrochloric acid solution (0.1M).
Elution can be performed repeatedly (several hundred times), with
a correspondingly decreasing activity, the generator can serve
for about 1-3 years. Positron gallium-68 is thus operatively and
long-term available for clinical use in PET at the workplace of
nuclear medicine.
The sequence 68Ge --> 68Ga is also used in closed "germanium" phantoms
for calibration and testing of PET gamma cameras (see "Phantoms and phantom measurements in nuclear medicine", section "Tomographic phantoms"). The source of 511keV
annihilation radiation, detected by a PET camera, is daughter
gallium-68 (formed by the interaction of
positrons emitted by 68Ga with electrons of the material),
the parent germanium does not participate in the radiation.
The actual parent isotope 68Ge is prepared by
irradiating gallium isotopes in a cyclotron in nuclear reactions 69Ga(p,2n)68Ge, 72Ga(p,4n)68Ge, or 66Zn(a,2n)68Ge, resp. by
fragmentation strip reactions (p, p-xn) by ejecting x- neutrons
from bromine or germanium isotopes.
Germanium 71 Ge
is converted to the ground state 71Ga by electron capture with a half-life of
11.4 days, a characteristic gallium X-ray is emitted. This
isotope was used in experiments with radiochemical detection
of solar neutrinos using the reaction 71Ga (n, e-) 71Ge. After
radiochemical separation of the germanium formed, the
characteristic X-rays and Auger electrons emitted from gallium
atoms after conversion by 71Ge electron capture was measured (§1.2,
part "Neutrinos - "ghosts" between particles", passage "Neutrino detections").
Germanium 76 Ge
was previously considered stable isotope. Later
experiments have shown that with an enormously long half - life
of 1,7.1021 years, it is transformed by double beta decay b-
b- to
selenium 75Se
(§1.2, part "Radioactivity beta-", passage "Double beta decay"
). .........
Selenium Se 34 is a gray-white semi-metallic element,
which occurs relatively rarely in nature (representation
in the earth's crust approx. 0.005-0.17 mg/kg). Its main use is in electronics due to its outstanding photoelectric
properties - photoelectric cells - and as a component of some
semiconductor materials. Selenium has 5 stable isotopes: 74Se (0.89%), 76Se (9.37%),
77Se (7.63%), 78Se (23.77%)
and 80Se (49.61%). Natural selenium also
contains 8.73% of the very long - lived radioactive isotope 82Se (T1/2
= 1.087.1020 years; its
radioactivity is negligible, it can be considered practically
stable). Of the radioactive isotopes, one
found practical application :
Selenium
75 Se
with a half-life of 119.8 days is converted by electron
capture (EC) to a series of excited states of arsenic 75As, predominantly to
an excited state with an energy of 401keV (95.9%); conversions to other higher excited states have a very
low intensity (<0.01%). During cascade
deexcitation, about 20 energies of gamma photons are emitted, of
which energies of 121, 136, 264, 279 and 401 keV have a more
significant intensity.
Conversion scheme and gamma spectrum of selenium 75Se. Apology: The semiconductor
spectrum was measured on an older Ge(Li) detector with degraded
properties.
Gamma peaks 121 (17%), 136 (58%), 264 (58%),
279 (25%) and 401 (11%) [keV] dominate in the gamma
spectrum 75Se.
Many other higher peaks in the range of 400-822keV are so low
(<0.01%) that in our weak sample (approx.
3kBq) they did not displayed in the
spectrum.
The isotope 75Se is prepared in a cyclotron by irradiating selenium (enriched in the isotope 74Se) with neutrons by the reaction 74Se (n, g) 75Se in a nuclear
reactor. It is used as a source of gamma radiation
in industrial radiographic applications (§3.3, section "Radiation
defectoscopy") as an alternative to the iridium 192 Ir described below. 75Se it has a longer
half-life compared to iridium and provides a softer spectrum of
gamma radiation (it may be suitable for
defectoscopic testing of thinner metal layers of about 2-3 mm,
where lower gamma energy provides better radiographic contrast).
Selenium-75 is rarely used in nuclear
medicine in the form of 75Se-selenelethionine, in which the
sulfur atom in the amino acid methionine is replaced by
a selenium-75 atom. After intravenous administration, the amino
acid metabolism of methionine and proteosynthesis
are examined. In the 60s-80s, this radiopharmaceutical was used
for scintigraphic diagnosis of the pancreas (intake of amino acids in the pancreas is a reflection
of the rate of synthesis of digestive enzymes - §4.9.3, passage
"Scintigraphy of the pancreas"). Also, 75Se-
tauroselcholic acid is rarely used to diagnose malabsorption
of bile acids, in the assessment of reduced absorptive
function of the terminal ileum (e.g. in Crohn's disease,
inflammatory, toxic or radiation damage).
For use in nuclear medicine, the disadvantage
of 75Se is
its too long half-life (120 days) ...
Rubidium
- Krypton -Xenon
Rubidium Rb37 is a light soft shiny metal from
the group of alkali metals. It is very reactive, so it occurs in
nature only in compounds (representation in
the earth's crust about 100-300 mg/kg). It has a single stable isotope of 85Rb
(72.17%), in the natural rubidium the remaining 27.83% is the primordial
radioactive isotope 87Rb (see below). Of the rubidium radioisotopes, three
isotopes with diametrically different half-lives are important :
Rubidium 81 Rb
With a half-life of 4.57 hours, it is converted by electron
capture (39%) and b+-radioactivity (25%) to excited states
of krypton 81Kr. The most common transformation to form the
metastable isomer 81mKr with an energy of 191keV and a short half-life of 13
seconds.
The artificially produced isotope 81Rb is used as the parent generator nuclide
for the preparation of the short-term metastable isomer 81mKr
for lung ventilation scintigraphy in nuclear medicine (see 81m Kr below). 81Rb is most often prepared by the reaction 82Kr (p, 2n) 81Rb by irradiation of
krypton (possibly enriched with the isotope
82Kr) with protons accelerated in a cyclotron.
Rubidium
82 Rb
With a half-life of 1.27 minutes is converted by b+-radioactivity
(96%) and electron capture (4%) to basal (87%)
and to excited states (mainly to the level
of 776keV - 8%) of krypton 82Kr. During
deexcitation, more gamma photon energies are emitted, only 776keV
energy is more significant. The gamma spectrum of rubidium-82 is
dominated (as with any positron
radionuclide) by the e-e+ annihilation
peak 511 keV (191%), the gamma peak 776keV (13%) and the
weaker 1395keV (0.5%) are also visible; only at high
magnification are other very faint peaks up to 3900keV visible.
.......picture.........
add.................. Spectrum ......
The short-term isotope 82Rb is obtained by
elution from a generator 82Sr/8 Rb, whose parent isotope 82Sr has a half-life of
25.4 days - see passage "Sr-82". Rubidium-82 is used experimentally for PET
scintigraphy of myocardial perfusion (§4.9.4., passage "Scintigraphy of myocardial perfusion"). It is applied in the form
of chloride 82RbCl, behaves as an analogue of potassium (similar to thallium 201 Tl ).
Rubidium 87 Rb
is a primordial radioisotope of rubidium (27.83% content of
natural rubidium). With a very long half-life of 4,81.1010
years, the b- radioactivity is converted to
strontium 87Sr in the ground state. 85Rb is used in isotope geochronology to
determine the age of minerals and rocks - rubidium-strontium
dating (see "Radioisotope dating" above).
Krypton Kr 36 is a chemically inert rare gas, in the
Earth's atmosphere it is found in a concentration of about 0.0001%, from which it is obtained by fractional distillation
of liquefied air. Used to fill light bulbs and discharge lamps.
It has 6 stable isotopes 78Kr (0.36%), 80Kr (2.29%), 82Kr (11.6%), 83Kr (11.5%), 84Kr (56.99%), 86Kr (17.28%).
From a number of radioactive isotopes, it has a practical
application of especially 81mKr in nuclear medicine and 85Kr in industrial applications.
Krypton
81m Kr
is a metastable nuclear isomer of krypton-81,
which with a short half-life of 13.1 seconds
deexcites to its ground state, from 65% of the 191keV
gamma photon emissions, from 35% by internal conversion to
envelope electron emissions. 81mKr is obtained from the generator 81Rb/81mKr.
The principle of this generator is in the left part of figure.
The parent rubidium 81Rb is fixed in the solid phase in a small column,
through which a stream of elution air is passed
by means of a fan (air pump with adjustable
power). Through the radioactive decay of rubidium-81, the
continuously released daughter gas krypton 81mKr is entrained by the passing air and led into a breathing
mask, from which the patient inhales a mixture of air
and radioactive 81mKr. One-way valves are included in the
circumference of the breathing mask, and a mixing valve for
outside air is also connected to ensure free breathing. Exhaled
air is led to the extinction vessel (volume approx. 30 liters), from
which, due to the very short half-life of 81mKr, practically non-radioactive air emerges (in contrast to the xenon 133 Xe below, which had to be
piped outside the building).
Note: As can be seen from
the decay scheme in the right part of the picture, after rapid
deexcitation of 81mKr the resulting krypton 81Kr in the ground state is not stable, but is weakly
radioactive (see below 81Kr), but due to the long half-life it it does not show
in any way (well below the level of the natural radiation
background).
During this
examination of pulmonary ventilation, inhaled
air with a trace content of radioactive 81mKr enters the pulmonary alveoli, and the
emitted gamma radiation of 191keV is scanned by a gamma
camera. The scintigraphic image of the area with reduced
activity shows areas of the lung with poor ventilation,
where krypton-81m, and therefore no air, does not get (either at
all or reduced) - see §4.9.5 "Lung scintigraphy
(nuclear pneumology)".
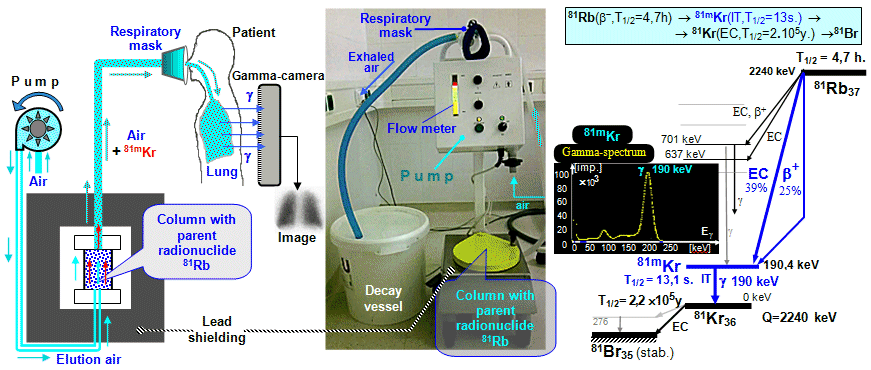
Generator 81Rb/81mKr and lung ventilation .
Left: Principle of generator operation
and scintigraphic examination of pulmonary ventilation. Middle:
One of the design arrangements of the Rb-Kr generator. Right:
Conversion scheme 81Rb and 81mKr; in the
black field is the scintillation spectrum of gamma radiation 81mKr.
Krypton 81
Kr
is transformed by electron capture to a stable 81Br with a long
half-life of 2.29.105 years. In 99.7%, 81Br is formed in the ground state, in 0.3% in the excited
state 276keV. It occurs in nature in very low trace amounts as a cosmogenic
radionuclide.
Krypton 85
Kr
With a half-life of 10.75 years,
is transformed by beta-
-radioactivity (Eb max = 687keV) to a stable 85Rb, in 99.7% to the
ground state, in 0.44% to an excited state of 514keV. During
deexcitation, gamma photons with the same energy of 514keV
(0.43%) are emitted, in trace amounts (0.0000022%) then during
cascade deexcitation of gamma 362.8keV and 151.2keV. In nature, 85 Kr is formed in very
low trace amounts as a cosmogenic radionuclide, but at
present a much larger amount comes from nuclear reactors
(beta transformations from the primary fission product 85As). Due to the
longer half-life, 85Kr accumulates in the fuel during reactor operation. A
small part of this krypton escapes directly from the reactor,
most of it during the reprocessing of nuclear fuel, from where it
is released into the atmosphere.
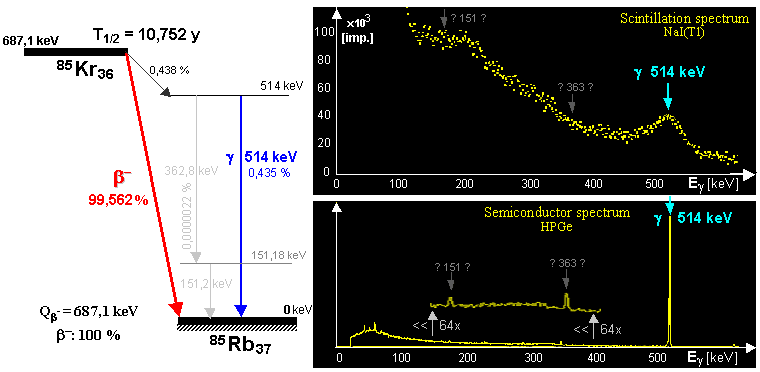
Conversion scheme and gamma-spectrum of krypton 85Kr
I apologize for the poorer quality of gamma spectra (statistical
fluctuations, high background). The spectrum was measured on a
lamp with a very low content of Kr-85 in the gas charge. Weak
peaks 151 and 363 keV are almost not visible in the natural
background (if I can get a stronger Kr-85 sample, the measurement
would be repeated) ...
Krypton 85Kr can be produced in a nuclear reactor by neutron
fusion of natural krypton-84: 84Kr (n, g) 85Kr; with formation of a metastable predominantly 85mKr to 4.48 with a
half hours deexcited to basic state 85Kr. In most cases, however, 85Kr is isolated from fission products of nuclear fuel by
the PUREX method.
Krypton 85Kr is used in technical practice as an additive to the gas
filling of lamps (approx. 10kBq/m3), where beta-electron ionization facilitates the
ignition of an electric discharge at a lower voltage (§3.7, section "Radioactivity in lamps"). Another use is as a tracer
gas radionuclide for detecting leaks in
hermetic vessels and pipes. Also in various other laboratory
applications ...
Xenon
is included in this section somewhat atypically , its proton number and position
in the periodic table of elements rank it only after iodine (and
before cesium). From our point of view, there are two reasons for
this atypical classification: 1. Xenon is an inert rare gas
similar to krypton; 2. The most important radioisotopes
of krypton ( 81mKr ) and xenon ( 133 Xe ) have their main uses in nuclear
medicine: for scintigraphic examination of pulmonary
ventilation
.
Xenon Xe 54 is a chemically inert rare gas,
in the Earth's atmosphere it is found in a concentration of about
5.10-6
%, from which it is obtained by fractional distillation
of liquefied air. It is used to fill light bulbs and especially
discharge lamps. It has 8 stable isotopes: 124Xe (0.1%), 136Xe (0.09%), 128Xe (1.9%), 129Xe (26.4%), 130Xe (4.07%), 131Xe (21.23%), 132Xe (26.91%), 134Xe (10.44%), 136Xe (8.86%).
Note: Of interest
is the above-mentioned stable isotope 129Xe , which is a daughter product of the
radioisotope 129
I and whose increased content
in some meteorites is used in the so-called iodine-xenon
chronometry, to determine the time of their formation - see
above section "Radioisotope (radiometric) dating", passage "Dating
using decayed radionuclides".
Of the radioactive isotopes of xenon, 133Xe is occasionally used :
Xenon 133 Xe
With a half-life of 5.25 days, is converted by b-
-radioactivity to 133Cs, in 99.1% to an excited level of 81keV, 0.87% to a
higher level of 160.6keV and only 0.0092% to an even higher level
383.8keV. During deexcitation, gamma photons with an energy of 81keV
(37%), then 79.6keV (0.29%) are emitted, with a very low
intensity then higher energies of 160.6keV (0.068%), 302.9keV
(0.0058%) and 383.8keV (0.0028%).
The isotope 133Xe is most often obtained by fission of uranium
(enriched in the isotope 235U) by neutrons in a nuclear reactor. Another possibility
is neutron irradiation of natural xenon in a nuclear reactor
using the reaction 132Xe (n,g)133Xe (there is also a metastable 133mXe, which with a
half-life of 2.2 days of gamma 233keV photon emission
isomerically deexcites to the ground state 133Xe), or neutron irradiation of
cesium by the reaction 133Cs (n, p) 133Xe.
Gas xenon 133Xe is used *) in nuclear medicine for the diagnosis
of lung ventilation function - it is described in more
detail in §3.11b "Dynamic lung scintigraphy (133-Xenon
ventilation)" of the book
"Comprehensive
evaluation of scintigraphy".
*) 133Xe was often used mainly in the 80's. Its disadvantages
were difficult availability, complicated handling of gaseous
xenon and the need for special spirometric breathing
apparatus . At this price, however, it allowed a very comprehensive
examination of respiratory function . Currently,
ventilatory lung scintigraphy is performed mostly with the 81m
Kr generator krypton
described above.
Metastable xenon 131m Xe
is described below in the section "Radioiodine
131
I", passage "Metastable
xenon 131m Xe".
Strontium, Ytrium
Strontium Sr 38 is a relatively soft gray-white shiny metal from the
alkaline group. It is highly reactive, in nature it occurs only
in compounds, in the earth's crust in the amount of 0.03-0.04%.
Strontium occurs in nature in the form of 4 stable isotopes: 84Sr (0.56%), 86Sr (9.86%), 87Sr (7%), 88Sr (82.58%).
Note : Isotope 87Sr in nature is formed by the beta-radioactivity of the
rubidium isotope 87Rb - it is radiogenic; long-term radiometric
dating is performed using the ratios of the isotopes 87Sr, 86Sr and 87Rb (see "Radioisotope
(radiometric) dating"
above).
Of the radioactive
isotopes of strontium, two are particularly important :
Strontium
89 Sr ,
with a half-life of 50.5 days is converted by b-
-radioactivity from 99.99% to the ground state of
yttrium 89Y
(Eb max = 1488keV) and from 0.01% to the
excited state of 909keV (during
deexcitation, very weak gamma radiation of this energy is
emitted). It is therefore a practically pure
beta emitter.
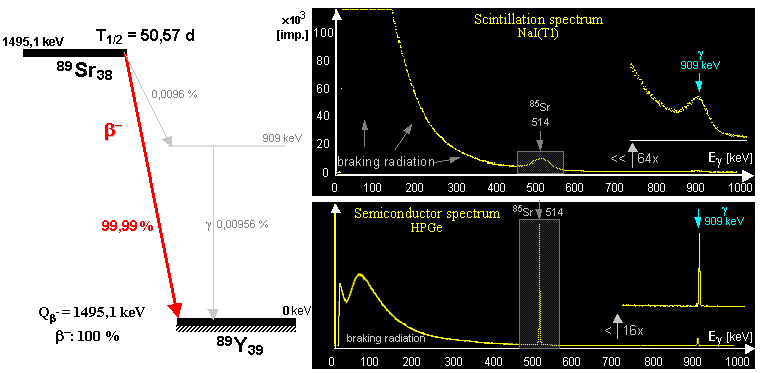
Conversion scheme and gamma spectrum of
strontium 89Sr.
Note: The gamma peak of 514 keV in the
middle of the spectrum does not belong to 89Sr,
but comes from a small (approx. 0.01%) radionuclide impurity 85Sr
in the measured sample.
In the gamma-spectrum of 89 Sr we see mainly braking radiation
with a continuous spectrum, arising from the interaction of fast
beta beta electrons with atoms in the sample material; with a
decreasing tendency it stretches up to an energy of about
1000keV. With a longer spectrometric acquisition, a faint gamma-ray
photopeak with an energy of 909keV can
be seen here when magnified.
When measuring our specific sample (" Metastron ") also
showed a peak 514 keV, originating from the radionuclide impurity
85 Sr (its origin is mentioned in the note below) .
Isotope 89Sr is prepared either
by irradiating strontium (enriched in the 88Sr isotope ) with slow neutrons by the 88Sr (n, g) 89Sr reaction in a nuclear reactor, or by irradiating
yttrium with fast neutrons in the 89Y (n, p) 89Sr reaction. It can also be radiochemically separated
from uranium fission products.
Note: Typical radionuclide impurities
in 89Sr
preparations are 85Sr (formed by the reaction of n,g with the isotope 84Sr contained in the
target) and 90Sr, arising from the isotope 88Sr, or abundant in
fission products.
Radioisotope 89Sr in the form of
chloride ("Metastron") is used in nuclear
medicine for the palliative therapy of bone metastases
of the ca prostate and breast (§3.6,
section "Radioisotope
therapy").
Strontium 90 Sr
with a half-life of 27.78 years converts b-
-radioactivity to the radioactive yttrium 90Y (Ebmax = 546keV) in the basic state.
Isotope 90Sr
is formed during the nuclear fission of uranium-235 and
plutonium-239 (with a yield of 5.7%) and is isolated from fission
products from spent nuclear fuel. Used in thermoelectric radioisotope
batteries (along with plutonium-238). An important use of strontium-90 is in 90Sr/90Y
generators for the preparation of yttrium 90Y
in nuclear medicine (see Y-90
below).
Strontium
82 Sr
- Sr / Rb generator
is converted to radioactive rubidium 82Rb
in the ground state by electron capture with a
half-life of 25.4 days. The strontium-82 isotope serves as the
parent isotope in the 82Sr/82Rb generator to obtain the short-lived
positron radionuclide 82Rb for use in PET positron emission
tomography (see passage "82Rb"
above).
Yttrium
Y39 is a
silvery gray metal similar to a group of lanthanides. In
terrestrial nature, it occurs only in compounds, in the earth's
crust in the amount of about 2-4 mg/kg. Yttrium is used to make luminiscence
substances. It has the only stable isotope 89Y. An important
radioactive isotope of yttrium is :
Yttrium
90 Y ,
which with a half-life of 64.1 hours is converted by b-
-radioactivity from 99.9885% to the ground state of 90Zr (Eb max = 2300keV) - is thus a
practically pure b- emitter.
Only in a small proportion of 0.017% there
are conversions to two excited states 90Zr 1760 (0.017%) and 2190 keV (negligible 0.0000014%).
The level of 2190keV transitions to the ground state by
gamma-deexcitation (gamma emission at the
limits of measurability). The level of
1760keV cannot be deexcited directly to the ground state, as it
is a monopole E0 junction 0+ ® 0+ (discussed in §1.2, section "Gamma radiation",
passage "Nuclear metastability and
isomerism"); there may be 2-g emission, internal conversion, or emission of e--e+
pair - 90Y
is therefore also a weak positron emitter.
The spectrum of 90Y gamma radiation is dominated by a continuous
curve of intense braking radiation,
which arises during the interaction of high-energy beta electrons
with the sample material (with a decreasing
tendency it stretches to an energy of almost 2MeV). In more detailed measurements with a longer exposure
time, it further appears in the spectrum peak
of weak annihilation radiation 511keV (0.006%) from
emitted positrons. The higher peaks 1761keV *) and 2186keV are
very weakly represented.
*) The 1761keV peak arises mainly by
simultaneous detection of both emitted gamma quanta at 2-g deexcitation of
the 1761keV level (direct gamma deexcitation is prohibited - E0);
therefore he is weak.
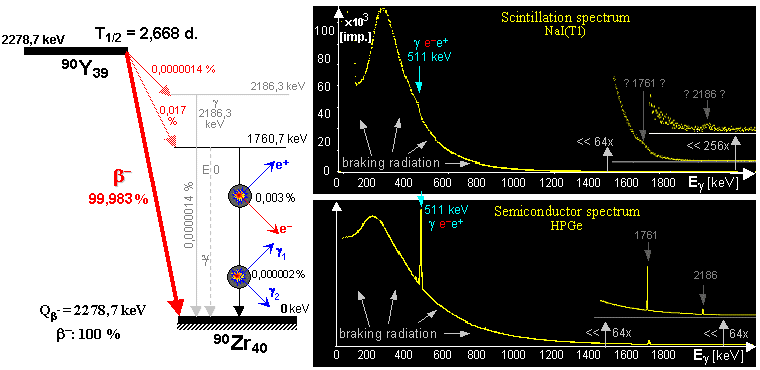
Conversion scheme and gamma spectrum of yttrium 90Y.
When measuring the spectrum of very weak gamma radiation, a
sample of yttrium was surrounded by a layer of 2.5 cm plastic for
shielding the beta hard electrons.
The Y-90 isotope is obtained mainly from the 90Sr/90Y
generator mentioned above. Alternative production
methods are irradiation of yttrium with neutrons in a nuclear
reactor 89Y
(n, g) 90Y (but has a low effective cross section), or using a cyclotron by irradiation yttrium with
deuterons 89Y (d, p) 90mY (IT, T1/2 =
3.2h.) ® 90Y, or rubidium by
alpha-particles 87Rb (a, n) 90mY (IT) ® 90Y.
The main use of the isotope 90Y, as a pure b-radiator, is in therapeutic
nuclear medicine (§3.6, part
"Radioisotope
therapy"), where high-energy beta-particles are used in radioimmunotherapy,
palliative therapy of metastases or radiation
synovectomy in larger joints. The relatively longer reach of
these beta electrons in the tissue allows (thanks to the "crossfire"
effect) the even irradiation of larger tumors, often showing
heterogeneous blood flow and hypoxia.
Weak positron and
annihilation radiation has no direct radiation significance, it
is completely overwhelmed by intense braking radiation. In
principle, however, even a weak 511 kV annihilation radiation can
be used to image biodistribution therapeutic
radiopharmaceuticals labeled with 90Y using positron emission tomography PET
*) with better resolution than single photon scintigraphy
(planar, SPECT) using bremsstrahlung. Another option for imaging
yttrium biodistribution is PET scintigraphy using the 86Y
positron yttrium mentioned below.
*) The coincidence detection mode strongly
suppresses the braking radiation, so that even weak annihilation
radiation can be successfully detected. The principle of PET is
described in §4.3, section "PET cameras".
Yttrium 86 Y
With a half-life of 14.7 hours, is converted by b+-radioactivity
(26%) and electron capture (74%) to strontium 86Sr at more than 20
highly excited levels. In addition to the e-e+ annihilation
peak 511keV (35%), the gamma spectrum of yttrium-86
contains a large number of gamma peaks from deexcitation of 86Sr levels - 443 (16%), 515+580 (5%), 628 (33%), 703 (15%), 777
(22%), 836 (5%), 1026 (10%), 1077 (83%), 1115 (31%), 1854 (17%),
1921 (21%) ) keV; at high magnification, a number of other weak
peaks of mostly high energies can be seen, up to 3800keV.
.........picture...... - ....... conversion scheme, spectrum
........
Isotope 86Y is prepared by proton bombardment of a strontium
target (highly enriched in the isotope 86Sr) in a cyclotron reacting the 86Sr (p, n) 86Y.
The yttrium-86 is
yet experimentally indicates diagnostic radiopharmaceuticals
for use in positron emission tomography PET as radiotracers
for accumulating pharmacokinetics and therapeutic
radiopharmaceuticals labeled with 90Y in the organism (isotope Y-90 is
a pure beta, its biodistribution is difficult to visualize
scintigraphically - via braking radiation).
It's a special example of teranostics (cf. §4.9, passage "Combination
of diagnostics and therapy - teranostics"), where in this case one
isotope (86Y)
of the same element serves as a diagnostic indicator of
biodistribution of another isotope (90Y) used in therapeutic radiopharmaceuticals - eg in 90Y-conjugate of
monoclonal antibodies.
Zirconium
Zr 40 is a silvery-gray metal from the group of transition
metals, very chemically resistant. In terrestrial nature, it
occurs only in compounds, in the earth's crust in the amount of
about 160-200 mg/kg. Zirconium silicate ZrSiO4 is diamond-like in hardness and
crystalline structure. Zirconium is mainly used in nuclear
reactors (fuel cell encapsulation), due to its chermic and mechanical resistance and low
effective neutron capture cross section (§1.3,
section "Nuclear reactors"). Zirconium-niobium alloys
have superconducting properties and are used in some
superconducting electromagnets.
Zirconium has 4 stable isotopes of 90Zr (51.5%), 91Zr (11.2%), 92Zr (17.1%) and 94Zr (17.4%). Natural zirconium also contains 2.8% of the
radioactive isotope of primary origin 96Zr, which is transformed by double beta decay with a
hugely long half-life of 3,9.1019 years. Of the radioactive isotopes of zirconium, one
has found application :
Zirconium
89 Zr ,
which with a half-life of 78.4 hours is
converted by electron capture (76.2%) and positron radioactivity
(22.8%) to a stable 89Y in the excited state, mainly with an energy of 909 keV
(in fractional percent electron capture
even to higher levels of 1744, 2530, 2567 and 2622 keV). In the gamma spectrum of zirconium-89 there is a
significant e-e+ annihilation peak 511 keV (46%) (as with any positron radionuclide) and an even stronger gamma peak 909 keV
(99%); in the area of higher energies, weak
peaks of 1621, 1658, 1713 and 1745 keV are visible only at high
magnification.
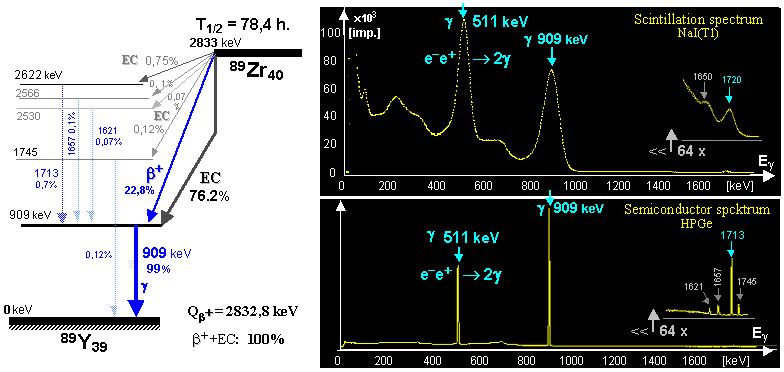
Conversion scheme and gamma spectrum of zirconium 89Zr.
The positron radionuclide Zr-89 is used in positron
emission tomography ("§4.3
"Positron emission tomography PET", §4.8 "Radionuclides and radiopharmaceuticals
for scintigraphy", section "Radionuclides and radiopharmaceuticals for
PET"). Due to its slightly longer half-life, 89Zr is suitable for
PET imaging of slower biological and physiological processes of
high molecular weight compounds, especially
radioimmunodiagnostics with bioconjugates of heavier monoclonal
antibodies (structure and
properties of monoclonal antibodies are discussed in more detail
in §3.6, part "Targeted biological therapy - monoclonal antibodies"), which have slower
pharmacokinetics, so that when labeled with short-term
fluorine-18 or gallium-68, radionuclides would not be sufficient
in time to be taken up in tumor foci.
Is test e.g. labeled
89Zr-cetuximab,
which blocks activation of the epidermal growth factor EGF-R,
which plays an important role in proliferation, differentiation
and survival of tumor cells. 89Zr-cetuximab may be used for PET diagnostic imaging of
cetuximab uptake in tumor and normal tissues prior to biological
immunotherapy. It can also be used theranostically
to detect the biodistribution of 90Y- or 177Lu-cetuximab in radioimmunotherapy.
Next, is tested the labeled 89Zr-trastuzumab, anti-epidermal growth
factor HER2, involved in proliferation,
angiogenesis, and metastasis of cancer cells. 89Zr-bevacizumab,
an anti VGEF antibody may display caused by
tumor angiogenesis, the vascular endothelial growth factor VGEF. A labeled 89Zr-J591 anti-PSMA monoclonal antibody
can be used to image PSMA positive prostate tumors.
Note: A
certain problem here may be the strong gamma-peak 909keV, whose
lower edge and Compton scattered radiation interferes with the
relatively wide working window of the analyzer of the measured
annihilation radiation 511keV (this may lead to a deterioration
of the image contrast due to false coincidences) ..?..
Molybdenum, Technetium
Molybdenum Mo42 is a relatively rare metal
element in terrestrial nature (approximately
1-7 mg/kg in the earth's crust). It has a number of applications,
especially in metallurgy. It occurs in a number of stable
isotopes: 92Mo (11.8%), 94Mo (9.3%), 95Mo (16%), 97Mo (9.5%), 98Mo (24.1%), 100Mo (9.6%) *).
*) Coincidence measurements with
semiconductor Ge detectors have shown that the 100Mo isotope is not
completely stable, but with a hugely long half-life of about 8.1018 years, it is
converted to ruthenium 100Ru by double beta decay.
Of the radioactive isotopes of
molybdenum, 99Mo is the
most important :
Molybdenum 99 Mo
is converted with a half-life of 66
hours by b-
-radioactivity to excited levels of the daughter nuclide
technetium 99Tc (left part of
the decay scheme). In 82% the conversion
takes place to a metastable excited level of 142 keV 99mTc
with a half-life of 6 hours, in 17% to a higher level of 921keV,
which emits g 740keV during deexcitation (via the level of 181keV). Molybdenum-99
is prepared in a nuclear reactor in two ways. The first method
consists in neutron irradiation of molybdenum (enriched in the isotope 98Mo) in the reaction 98Mo (n, g) 99Mo. More often,
however, 99Mo
is produced by fission of uranium (enriched in the isotope 235U) with slow neutrons, where one
of the fission products is 99Zr, from which the desired molybdenum-99 is formed by
two subsequent b-
-conversions: 99Zr(b-, T1/2=2,1s.)®99Nb(b-, T1/2=15s.)®99Mo.
Molybdenum 99 Mo serves as the parent radionuclide for obtaining
technetium 99mTc in a Mo-Tc generator (see below).
Technetium
The chemical element technetium
Tc 43 is practically non-existent in nature *), as it has
no stable isotope (with half-lives
of the most stable radioisotopes of technetium 97.98.99Tc - 2.6.106 years, 4.2.106 years, 2.1.105 years - they are not
long enough to preserve primordial technetium from a supernova
explosion to this day, as is the case with thorium, uranium or
potassium-40). Absolutely trace amounts of
technetium are formed during the spontaneous fission of
uranium-235. In the last few decades, however, a relatively large
amount of technetium has been generated during the operation of nuclear
reactors (§1.3 "Fission of
nuclear nuclei", section
"Fission products").
*) In Mendeleev's periodic table, the space
for atomic number 43 remained empty for a long time. The first
isotope of this element was discovered in 1937 during irradiation
of molybdenum with accelerated deuterons in a cyclotron of 99Mo (d, n) 97Tc.
This gave rise to the name "technetium"
as an element, whose isotopes can only be prepared
by an artificial - technical -
way.
Technetium
99m Tc
The most important radionuclide for nuclear medicine
is metastable technetium 99mTc (T1/2 = 6 hours), which is a pure gamma emitter
with energy Eg = 140 keV (88.5%). With negligibly low
intensity, g photons of 2.2 keV, 90keV, 142keV, 233keV and 322keV are
emitted (see below). However, conversion and Auger electrons,
produced from the atomic shell during the internal conversion of
deexcitation transitions, are relatively abundant (the mechanism is described in §1.2, passage "Internal conversion of gamma and
X-rays"). These are mainly low-energy electrons 1.6-2.9keV
(210%), 14-21keV (2.2%) and also electrons of medium energies
120-140keV (21%). During the internal conversion, jumps between
electron levels in the envelope also result in soft characteristic
X-rays of 18-21keV (8%).
99mTc is obtained almost exclusively by the above-mentioned
beta-decomposition of molybdenum 99Mo (T1/2 = 66 hours) in the so-called Mo-Tc generator.
However, it can also be produced directly ("instant" technetium) by proton bombardment of natural
molybdenum (enriched in the isotope 100Mo) in a cyclotron using a reaction of 100Mo (p, 2n) 99mTc - used only rarely.
Mo-Tc generators are
mostly of the elution type. Molybdenum 99Mo is absorbed on a
support (mostly Al2O3)
in an " insoluble" oxide form in a
"chromatographic" column. After the radioactive
conversion of the 99Mo core to the 99mTc daughter core , the resulting technetium atom is
released from an insoluble bond; combines with 4 oxygen atoms to
form the anion 99mTcO4-
pertechnetate. This daughter product is soluble
in water, whereby it can be separated from the starting
molybdenum by washing with water - elution (in
the picture on the left). Since the elution is performed with physiological
saline containing a NaCl salt, the pertechnetate anions are
immediately ionically bound to sodium to form sodium
pertechnetate Na 99mTcO4 -. In
this chemical form we obtain technetium from the elution
generator.

Elution 99Mo
/ 99mTc generator.
Left: Principle functional diagram of
the elution generator. In the middle:
Technical design of a sterile generator with an evacuated elution
vial.
Right: Conversion scheme of molybdenum 99Mo to technetium 99mTc, deexcitation to 99Tc and slow
transformations to stable ruthenium 99Ru.
New types of sterile elution
generators use an evacuated elution vial, into
which, after being "injected" by atmospheric vacuum,
the saline solution is automatically sucked through a tube
leading from the storage vial through the sorption column of the
generator with 99Mo (in middle part of figure). Within a few tens of
seconds, the vial is filled with 99mTc eluate.
A detailed decay scheme of 99mTc
is below in the right part of the picture. Default metastable
level 142keV (produced after beta-conversion of
the 99Mo)
isomerically passes first at the level of 140.5 keV, from where
it emits primary gamma rays with an energy 140.5
keV. With a very small proportion of 0.02%, there is a
direct deexcitation to the ground state, in which energy of 142.7
keV is emitted. Photons of very soft gamma radiation of 2.17 keV
are practically not observed, as they are almost 100% subject to internal
conversion. The core of technetium 99Tc in the
ground state (after the isomeric transition
from 99mTc) is beta-radioactive
and with a very long half-life of 200,000 years it is slowly
converted to stable ruthenium 99Ru. In a very small
percentage, there is a direct beta-conversion of 99mTc
from a metastable level *) to 99Ru (the basic state of the 99Tc nucleus is
"bypassed") - mainly to excited levels 322 and 90 keV 99Ru.
Their deexcitation produces g-
radiation with energies of 322, 232 and 90
keV, but a very small representation. At 99mTc
radioactivity, soft characteristic X-rays with
energies of »2-3keV (L-series) and »18-22keV (K-series) are
also emitted, as well as a larger number of low-energy conversion
and Auger electrons (up to 4 electrons/1 conversion), mostly
with energies »1.6-3 keV, smaller amounts »120-140 keV (mentioned
above).
*) In this process, the commonly stated
energy 142keV of this metastable level with respect to the ground
state of 99Tc is not applied , but the energy of
436keV measured with respect to the ground state of the daughter 99Ru.
This is a special case of branched transformation,
discussed above in the "Conversion
Schemes" section.

Detailed conversion scheme of technetium 99mTc - an
important radionuclide in nuclear medicine.
Left: Formation of metastable 99mTc
by beta-radioactivity of molybdenum 99Mo. Right:
The metastable level of 142 keV, in addition to the
dominant deexcitation to the ground state of technetium 99Tc,
can with low probability by beta- -radioactivity
convert directly to excited levels of ruthenium-99 .
In the standard 99mTc scintillation
gamma spectrum (on a scintillation spectrometer or gamma camera)
we observe only one significant photopeak of 140keV
energy - in the picture on the left (on a semiconductor spectrometer we can distinguish a
weak line of 142.7keV with careful long-term measurement). Weak peaks from the excited levels of 99Ru,
arising from 99mTc by "bypass" of 99Tc,
especially 322keV, can be seen spectrometrically only after
filtering out the strong radiating line 140keV with a layer of
about 4-5mm lead; the 740keV of (possible) contaminant 99Mo line is also
visible on this filtered spectrum - right part of the figure.
Note :
Line 90keV, which is stronger than 322keV, we do
not see in this filtered spectrum, because it is completely
absorbed by the lead layer.
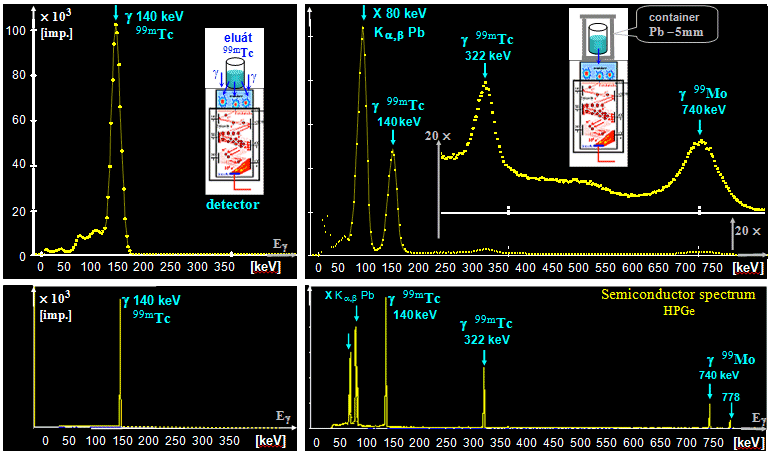
Gamma-spectrometric measurement of the 99mTc eluate (top - scintillation
spectrum, bottom - semiconductor
spectrum).
Left: Basic gamma radiation
spectrum 99mTc. Right: Filtered spectrum
of gamma radiation measured through the shielding layer of a 5 mm
lead container, in which a very weak g -line 322keV of 99mTc and a gamma line
740 and 778 keV of the contaminant 99Mo can be observed .
Technetium-labeled radiopharmaceuticals (§4.8 "Radionuclides and
radiopharmaceuticals for scintigraphy") are widely used
in static+SPECT and dynamic scintigraphy of
kidneys, liver, lungs, heart, brain and other organs, as well as
in tumor diagnostics (§4.9 "Clinical
scintigraphic diagnostics in nuclear medicine").
Indium, Tin
Indium In 49 is a soft, easily fusible metal (157
°C) of light color, relatively rarely
present in nature (approx. 0.1 mg/kg in the
earth's crust). It is used for the
preparation of low-melting alloys, mirror plating, in electronics
in the production of optoelectronic components. It has two stable
isotopes, 113In (4.3%) and 115In (95.7%).
Tin Sn 50 is also a soft, easily fusible metal (232 °C) of light color, somewhat
more abundant (approx. 2-4 mg/kg in the
earth's crust). It has a number of stable
isotopes: ........
Of the radioactive
isotopes of indium and tin, three radionuclides are of some
importance :
Indium
111 In
It is converted with a half-life of 67.4 hours by electron
capture to cadmium 111Cd in the excited state of 416keV, during the cascade
deexcitation of which gamma radiation with energies of 171 and
245 keV is emitted (decay scheme in the
left part of Fig). The 111In gamma spectrum
consists of two significant photo peaks of the mentioned energies
(spectrum in the right part of the picture); when measured with high detection efficiency, a weaker
summation peak around the energy of 416keV is also
visible in the spectrum. 111In is produced by irradiating cadmium (enriched in the isotope 111Cd) with accelerated protons in a
cyclotron by the reaction 111Cd (p, n) 111In.
In nuclear medicine 111In quite often used for scintigraphy of tumors and
inflammatory foci, for immunoscintigraphy and scintigraphy of
liquor pathways (§4.9 "Clinical
scintigraphic diagnosis in nuclear medicine", "Scintigraphy of liquor pathways"...).
Conversion scheme and gamma spectrum of indium 111In.
T i n 113
Sn
With a half-life of 115 days, it is converted by electron capture
to two excited states of indium 113In: in 98.2% the excited state of indium 113mIn
is formed, in 1.8% the level of 647keV, which also passes to the
level of 113mIn by emission of the gamma photon 255keV. The
radionuclide 113Sn is produced by irradiating tin (enriched in the
isotope 112Sn)
with neutrons in a nuclear reactor using the reaction 112Sn (n, g) 113Sn. This
radionuclide serves as the parent isotope for obtaining indium 113mIn in the generator
113Sn/113mIn.
Indium 113m
In
Metastable 113mIn with a half-life of 1.66h. deexcites to the ground
state of gamma photon emissions with an energy of 392keV. It is
obtained from the above-mentioned generator 113Sn/113mIn. In nuclear medicine, it was used in the 1960s -1980s
for similar scintigraphic methods as now 111In, as well as for examination of the skeleton, brain or
kidneys. Later, it was gradually displaced by
radiopharmaceuticals labeled with 111In and 99mTc, whose longer half-lives and lower gamma energies are
more suitable for scintigraphy.
Iodine
Element iodine I 53 of the halogen is highly reactive and
occurs in nature only in compounds; is
relatively rarely present (approx. 0.1-5
mg/kg in the earth's crust, in sea water approx. 0.6 mg/l). In the
solid state, they form dark purple leaf crystals, which sublimate
directly into the gas phase at atmospheric pressure. Iodine is one of the biogenic elements,
it is used mainly by the thyroid gland for the
production of hormones, especially thyroxine. It has a number of isotopes in
the range 108I - 144I, of which only one 127I is stable. Iodine is the element with
the largest number radioisotopes
used in practice, especially for applications in nuclear
medicine; we will mention them here in order of
importance :
Radioiodine
131 I
The most important radioisotope of iodine is radioiodine 131I
(T1/2 = 8 days, b- with max. energy 606keV, main energy g 364keV).
Radionuclide 131I is converted (according
to the decay scheme in the figure on the left) by b- -radioactivity to excited states of
the daughter nuclide xenon 131Xe, which is already stable (non-radioactive). The dominant "channel" of beta-conversion is
to an excited level of 364.5 keV (89%), which in
81% deexcites to baseline 131Xe a in 6% deexcites to a level of 80keV (which then
deexcites to baseline). In 2% the conversion occurs to the
excited level of 722keV, in 7% to the level of 637keV, in
fractions of a percentage to some other excited levels (including the interesting metastable
level 164keV of the daughter isomer 131mXe - see below "Metastable
131m Xe").
Conversion scheme and gamma spectrum of radioiodine 131I.
Note: This spectrum was measured with a
"fresh" iodine sample in which the metastable xenon 131mXe
had not yet accumulated .
In the scintillation and
semiconductor spectrum of gamma radiation 131I is dominated the main photopeak capturing the energy
of radiation g 364keV. Towards higher energies, two
weaker peaks, 637 and 723 keV, are visible. In the region of
lower energies we also see weaker peaks 284 and 80 keV, at the
very beginning of the spectrum then the characteristic X-ray Ka,b of
xenon 30keV (low-energy lines La,b
4-5keV on a common gamma detector not visible).
Metastable
131m Xe
With beta-radioactivity of radioiodine 131I, one of the excited levels of daughter 131Xe is a metastable
131mXe level with an energy of
164keV, which deeexcitates to the ground state of the 131Xe nucleus with a
half-life of T1/2 = 12 days; this excited metastable state can
therefore be considered as a separate radionuclide 131mXe.
Only 0.38% of 131I decays occur at this metastable level, and in
addition, its deexcitation is strongly subject to internal
conversion (from 98% ),
so only about 0.021% is emitted as 164keV gamma radiation (see
below). In a hermetically sealed preparation of 131I, a radioactive
equilibrium between the dynamics conversion of 131I and 131mXe is reached after
about 14 days from the production of the 131I isotope (however, the gaseous
daughter xenon continuously escapes from the open sample). It can be said that each preparation of
radioiodine-131 is a mixture of two radioisotopes:
the parent beta-gamma-radionuclide 131I with a half-life of 8 days and the daughter metastable
gamma-radionuclide 131mXe with a half-life of 12 days.
The faint 164keV photopeak from metastable
131mXe is
not visible in the scintillation spectrum of the sample 131I, because it lies
in the Compton main energy scattering region of 364keV (interferes with the backscatter peak). Even at the semiconductor spectrum, it is not obvious
at first glance, it is visible only after a strong enlargement of
the spectrum section between 100-200 keV. For a more accurate
spectrometric measurement of metastable 131mXe, we devised a small "trick" consisting of
the following simple experiment (easily
performed in a nuclear medicine workplace) :
We used a liquid sample of 131I
in the form of NaI iodide with an activity of about 100MBq (radioiodine commonly used for thyroid therapy), hermetically sealed in the vial for
about 14 days. We then aspirated air above the
surface of the sample, containing daughter xenon gas, into a
syringe with a needle; we closed this syringe immediately. In
addition to air, the gaseous sample in the syringe thus obtained
will also contain daughter xenon-131 containing
metastable 131mXe, whose gamma radiation can be
measured in a spectrometer - we thus got rid of the interfering
parent isotope 131I *), from which we separated the
daughter 131mXe. The resulting sample (with an
activity of about 3 kBq) for spectrometric
measurement was then prepared by transferring about 1/2
milliliter from the aspirated gas charge in the syringe to a
small measuring cuvette.
*) If the gaseous filling contains
evaporated gaseous iodine, we can get rid of it by shaking a
solution of inactive potassium iodide or sodium hydroxide. In our
measurement, however, this was not necessary, the aspirated
sample was completely pure, without the
slightest trace of iodine 131.
This measurement also shows that iodine does
not evaporate from the sodium iodide solution , because
the iodine atoms are bound by a strong ionic bond with sodium
atoms. Thus, it is not a true the paradigm that
when working with open iodide Na 131I, the
radionuclide iodine-131 is released ("sublimed") into
the air. Only the gaseous radionuclide xenon 131mXe
can be released. The situation is different for more complex
radioiodine compounds, eg 131I-MIBG, where the binding
is not so strong and a certain smaller amount of iodine is
released from the compound. In addition to xenon-131m, we can
also observe radioiodine-131 in the vapors above the sample
(spectrometrically demonstrated).
 |
Conversion scheme and
gamma spectrum of metastable xenon 131mXe.
A gaseous sample of 131mXe
was obtained by separation from a 131I
radioiodine preparate by aspirating the gas above the
level of a 131I sodium iodide solution. |
According to the transformation
scheme in the figure on the left, metastable 131mXe with a half-life
of 11.96 days deexcites by isomeric
transition (IT) to the ground state of 131Xe. In less than 2%,
it is a direct emission of gamma photons with an energy
of 164keV. In 98%, there is an internal
conversion (IC) of xenon atom sheath electron emissions,
mainly from the K shell (61%), partly also from the L shell (28%)
and from higher M, N shells (8%). The conversion
electrons emitted in this way then have a number of
discrete energies in the range of 129-163.8 keV. Electrons from
higher levels immediately jump to the "emptied places"
after the conversion electrons, which leads to an intense
emission of photons of characteristic X-rays
with discrete energies in the range of approx. 29-34keV. The
partial internal conversion of this X-ray also emits Auger
electrons with energies of 23-34keV (and also with very low energies of 2.5-5.5keV).
In the figure on the right, the resulting
gamma spectrum is measured with the 131mXe sample thus separated. The characteristic X-rays
(lines Ka,b) of 30keV xenon, arising due to strong internal
conversion during deexcitation of the excited level of 164keV,
dominate here. The photopeak of the 164keV 131mXe gamma line itself is significantly lower, but it is
very clearly visible in the spectrum, especially in the enlarged
section.
Production and use of radioiodine 131
I
Isotope 131I
can be obtained by irradiating tellurium with neutrons in a
nuclear reactor by reacting 130 e (n, g) 131mTe, with subsequent radioactive transformations 131mTe(g, T1/2=30hours)®131Te(b-, T1/2=25min.)®131I. In most cases, however, radioiodine 131I is prepared by
separation from fission products 235U.
Iodine 131I is of key importance in nuclear medicine for the
diagnosis (gamma component) and especially the therapy
(beta component) of thyroid disease (§4.9.1 "Thyrological
radioisotope diagnostics"
and §3.6, part "Radioisotope
therapy"). It is also used for targeted radioisotope therapy of
neuroendocrine tumors (131I-MIBG) and lymphomas (131I-Bexxar).
For scintigraphic diagnostics, 131I has a disadvantage
in the relatively high radiation exposure caused
by irradiation of the tissue with energetic beta particles.
Therefore, where iodine is essential, 131I has recently been replaced by the 123I isotope for
diagnosis (see below). In a number of examinations, even better,
eg 99mTc,
which is a pure gamma emitter with even much lower radiation
exposure.
Iodine
123 I
Another radioisotope of iodine used in nuclear medicine is 123I.
With a half-life of T1/2
= 13.2 hours, it
is converted by electron capture to tellurium 123Te *), mainly to its
excited level 159 keV (97%), from which comes
the main radiation energy g used for scintigraphy. Gamma radiation emitted during
dexcitation from higher energy levels (approx. 400-1000keV) has a
very low intensity, so it is practically not applied.
Deexcitation transitions in the atomic shell after electron
capture further produce intense characteristic X-rays
(Ka,b) of 27-31 keV.
*) Daughter tellurium-123 is weakly radioactive,
transformed with a very long half-life of 12 billion years by
electron capture to antimony (stibium) -123. However, the
activity of 123Te in the used preparations (tens
of MBq) after the disintegration of 123I is immeasurably
low, a million times smaller than the natural
background.
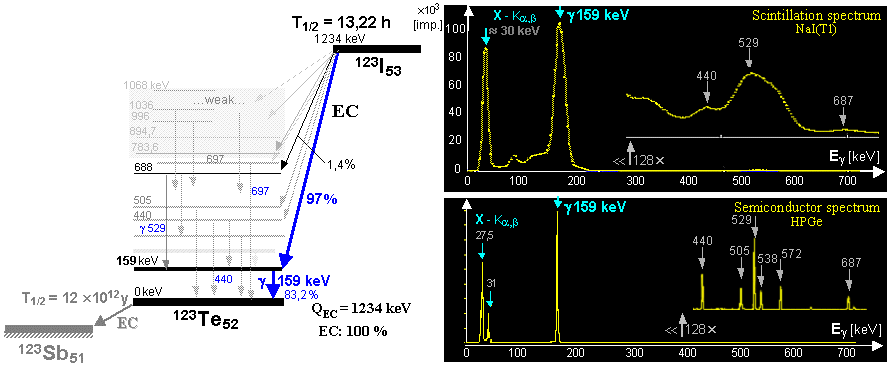
Conversion scheme and gamma spectrum of iodine 123I
Isotope 123 I is prepared by proton or deuteron irradiation of
tellurium or xenon in a cyclotron by several nuclear reactions.
With tellurium (enriched in the appropriate
isotope) it is by reactions 123Te (p, n) 123 I, 122Te (d, n) 123I, or 124 Te (p, 2n) 123I. A more common
method of producing the isotope 123I is to irradiate xenon (highly
enriched in the isotope 124Xe) protons with an energy of 25
MeV, in which three types of nuclear reactions take place
simultaneously: 124Xe (p, 2n) 123I - direct formation of 123-iodine; 124Xe(p,pn)123Xe(b+,EC, T1/2=3,9min.)®123I ; 124Xe(p,2n)123Cs(b+,EC, T1/2=5,9min.)®123Xe(b+,EC, T1/2=2,08h..)®123I. After irradiation from the target chamber with xenon
in approx. 12 hours (until 123Xe decomposes to 123I) the resulting 123I is separated and the remaining xenon 124Xe is recycled for
reuse.
By iodine-123 indicates some
radiopharmaceuticals for scintigraphy. It is mainly
sodium iodide Na123I for thyroid scintigraphy (§4.9.1
"Thyrological radioisotope
diagnostics"). 123I-ioflupane and 123I-IBZM are used for scintigraphy of receptor systems in
the brain (§4.9.8, section "Scintigraphy of receptor
systems in the brain"). Compared to iodine 131I, 123I has a more suitable gamma energy, and since it does
not emit beta radiation, it causes a significantly lower
radiation exposure for the patient during the
examination.
Iodine
125 I
Radioiodine 125I (X 27+31 keV, g 35keV) with a half-life T1/2 =
59.4 days is converted by electron capture
to an excited level of 35.5keV tellurium 125Te. Deexcitation to the ground state emits gamma
radiation of energy 35.5 keV (6.63%). After
electron capture, photons of characteristic X-rays
of tellurium Ka 27.2-27.4 keV (100%) and Kb 30.9-31.8 keV
(25%) are also emitted; this X-radiation is dominant. In
addition, after electron capture, due to the internal conversion
of gamma and X, a larger amount of Auger and conversion electrons
with energies of 0.7-30 keV (approx. 20
electrons /1 decay) is emitted from the
electron shell of the daughter tellurium. The 125I gamma spectrum
shows characteristic X-ray peaks around 30keV and a 35keV gamma
peak (they merge into one wider peak on the
scintillation detector) and, with good
detection efficiency, also a summation peak around
65keV, resulting from coincident detection of gamma + X photons
*). When measuring 125I samples in a well scintillation detector with a
detection efficiency of about 80%, the summation peak may even be
dominant.
*) During electron capture, there is a jump
of electrons in the envelope with the formation of a photon
characterized by X-rays and practically at the same time
deexcitation of the excited level of 35.5 keV 125Te with the emission
of the gamma photon. Both of these photons then enter the
detector at the same time and can be coincidentally
detected with the formation of a pulse corresponding to
the sum of both energies, about 65keV.
Conversion scheme and gamma spectrum of radioiodine 125I.
Isotope 125I is prepared by irradiating xenon (enriched
in the isotope 124Xe) with neutrons in the reaction 124Xe (n, g) 125Xe followed by radioactive conversion 125Xe (EC, T1/2 =
16.9 hours) ® 125I.
Iodine 125I is used in nuclear medicine for in vitro radioimmunoassay
(§3.5 "Radioisotope
tracking methods") and in radiotherapy for permanent interstitial
brachytherapy (§3.6, section "Brachyradiotherapy").
iodine 129 I
With a long half-life T1/2 = 1.57.107 years, is converted by beta- -radioactivity to an excited state of 39.6
keV xenon 129Xe, during the dexcitation of which gamma-photons
of the same energy are emitted. Due to the high internal
conversion of gamma photons, conversion electrons and strong
characteristic X-rays with an energy of around
30-34keV are also emitted (but still not as
intense as in the electron capture of iodine-125).
129I it also occurs in
trace quantities in nature - it is formed in the earth's crust
during the fission of uranium, in the atmosphere it is formed as
a cosmogenic radionuclide from xenon by reactions caused by
cosmic radiation. More recently, 129I is formed during the fission of uranium-235 and
plutonium-238, from where it is released during the reprocessing
of spent nuclear fuel. Due to the similarity of the gamma spectra
of I-129 and I-125, the 129I standards are used in the calibration
of detection systems for measuring 125I samples in radioimmunoassay.
Note: It is interesting to use the radioisotope 129I
, resp. analysis of the increased content of its stable daughter
isotope 129Xe in some meteorites, in the so-called
iodine-xenon chronometry for
determining the time of their formation - see above the section
"Radioisotope
(radiometric) dating", passage
"Dating using
decayed radionuclides".
Conversion scheme and gamma-spectrum of radioiodine 129I.
The spectrum is very similar to the
spectrum of iodine 125I shown above. Small diference is in the somewhat smaler
relative proportion of the characteristic X-ray with respect to
the gamma line 39.6keV. Furthermore, the summation peak X (Ka,b ) + g is not present,
because the characteristic X-radiation arises here due to the
internal conversion of gamma photons, which thereby disappear and
instead the emitted photons X have nothing to
"coincide" with. With each 129I radioactive conversion, either a 39.6keV gamma photon
or an X 30-34keV photon is emitted, never both at the same time.
Iodine
124 I
is a positron radionuclide that, with a
half-life of 4.18 days, is converted by beta+-radioactivity
(23%) and electron
capture (77%)
to the ground state (27%) and a number of excited states of 124Te: 603 (30%), 1325
(6%), 2295 (19%) keV and others. The relatively complex gamma
spectrum 124I is dominated by the annihilation e-e+ peak 511 keV (46%), then the gamma
peaks 603 (63%), 723 (10.4%), 1326 (1.5%), 1376 (1.7 %), 1509
(3.1%) and 1691 (11%) keV. A number of other weak gamma peaks (up to almost 3000keV) it is
visible only at high magnification. At the beginning of the
spectrum, weaker Ka,b peaks of 27-31 keV
characteristic X-ray tellurium are visible due to electron
capture. For applications of 124I, only positron radiation and the
resulting annihilation g- radiation
of 511 keV are important.
Conversion scheme and gamma spectrum of iodine 124I
Isotope 124I is prepared in a cyclotron by irradiating tellurium
isotopes with protons or deuterons in reactions 124Te (p, n) 124I, or 124Te (d, 2n) 124I, or 126Te (p, 3n) 124I.
Iodine-124 labeled
radiopharmaceuticals are used experimentally in PET
positron emission tomography ("§4.3
"PET positron emission tomography", §4.8 "Radionuclides and radiopharmaceuticals
for scintigraphy", passage "Radionuclides and radiopharmaceuticals for
PET"). In nuclear thyrology, Na124I can use when scintigraphy in the thyroid gland,
where due to higher sensitivity and resolution of PET compared to
planar and SPECT scintigraphy, 124I-PET/CT can improve the detection of metastases and
residual thyroid tissue, determination of tumor metabolic volumes
and pre-therapeutic dosimetry in radioiodine therapy, accurate
measurement iodine intake during treatment.
Due to the relatively longer
half-life (compared to eg 18F or 68Ga), 124I may be suitable for PET imaging of slower biological
and physiological processes of high molecular weight compounds.
It is especially radioimmunodiagnostics with labeling of heavier
monoclonal antibodies with slower pharmacokinetics.
It is tested
marked 124I-MIBG (meta-iodobenzylguanidine) for imaging neuroblastoma and pheochromocytoma (with possible radionuclide therapy
131I-MIBG). It
tested further the 124I-beta-CIT (2.beta.-carbomethoxy-3beta-
(4-iodophenyl) tropane) for PET imaging of
the dopamine transporter in the brain with Parkinson's disease.
Labeled iodouracyl 124I-2´-deoxyuridine (UdR), through its incorporation into
DNA and RNA molecules, shows the potential for tumor growth.
There are also considerations about the
therapeutic use of Auger electrons from
124I (is
discussed in §3.6, section "Radioisotope therapy",
paragraph "Beta and alpha radionuclides for
therapy", passage "Auger
electron therapy").
Note: A certain problem of 124I for PET
applications is the relatively low proportion of required
positrons (23%) and, conversely, the undesirable intense gamma
radiation 603keV (63%) often at the same time, which is
energetically close to the annihilation radiation of 511 keV - in
a relatively wide working window of the analyzer is practically
indistinguishable from 511 keV, which can lead to deterioration
of image contrast due to false coincidences. Furthermore,
the considerably high energy of the emitted positrons around
2100keV, which therefore have a relatively long
range of about 5mm, slightly impairs the spatial resolution of
the PET image.
Xenon Xe 54
, with its radioactive isotope 133 Xe, has been included above
in the "Rubidium-Krypton-Xenon" section, the "Xenon" passage, due to the similarity of the
applications. Metastable xenon 131mXe was described above in the section "Radioiodine
131 I",
passage "Metastable xenon 131mXe".
Cesium
Cesium Cs 55 is a light soft silvery yellowish alkali metal, easy to
melt (melting point 28.4°C, boiling point
670.8°C), which is very reactive, so it
occurs in nature only in compounds, and relatively rarely (about
1-5 mg/kg in the earth's crust). Cesium metal has low energy for
electron emission. In atomic and nuclear physics, it is therefore
used in photoelectric components, such as photomultipliers
- the photocathode is usually an alloy of cesium and antimony (§2.4 "Scintillation detection and gamma-ray
spectrometry", part "Photomultipliers"). The hyperfine structure
of electrons in cesium atoms is used in very precise so-called atomic
clocks (passage "Exact - ideal - measurement of space and
time" in §1.6
"Four-dimensional spacetime and special relativity" in
monograph "Gravity, black holes and space-time physics").
Cesium has a single stable isotope 133Cs.
Very important is the radioactive isotope of cesium :
Cesium
137 Cs
One of the best known and most widely used radionuclides in
general is cesium 137Cs. It is a b- + g emitter with a
single gamma radiation energy of 662 keV. With a
half-life of 30.05 years, 137Cs is converted by beta- -radioactivity to stable barium 137Ba - in 5.6% to the
ground state, in 94.4% to an excited level of 662keV (it is worth noting that the level of 662keV is metastable
with half-life 2.55 min., which is, however, completely
negligible due to the basic half-life of 137Cs 30 years). 137Cs is
used as the main standard for gamma-spectroscopy
(§2.4, part "Gamma radiation spectrometry"), further for irradiation in radiotherapy (§3.6, part "Isocentric
radiotherapy"), in defectoscopy (§3.3, part
"Radiation
defectoscopy") and in a number of other measuring and technical
applications.
137Cs is formed during neutron fission of uranium 235U (or plutonium 239)
in a nuclear reactor from the primary fission product of iodine 137I by two
transformations beta- : 137I(b-, T1/2=23s.)®137Xe(b-, T1/2=3,9min.)®137Cs; the yield is 6%. 137Cs preparations are obtained by radiochemical separation
from fission products (PUREX
method - §1.3 "Nuclear reactions and nuclear energy",
passage "Nuclear wastes").
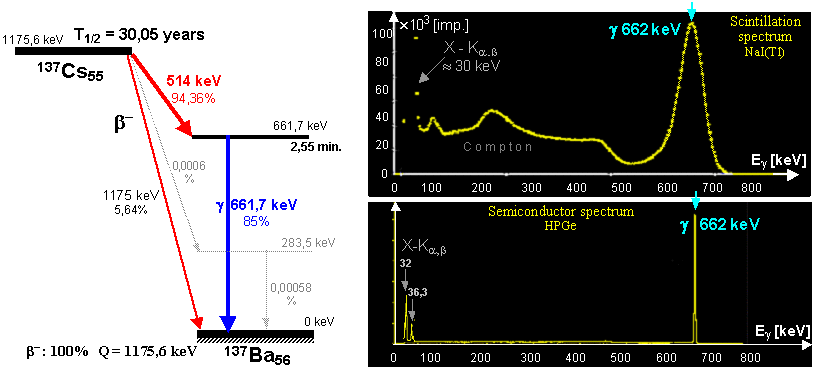
Conversion scheme and gamma spectrum of cesium 137Cs.
Barium
Barium Ba 56 is a soft light metal from the group of alkaline
earths, which is very reactive, so it occurs in nature only in
barium compounds, and relatively rarely (approximately 0.02-0.04%
in the earth's crust). Soluble barium salts are highly toxic. An
important mineral compound of barium is barium sulphate
BaSO4
called barite, whose aqueous suspension has a
relatively high density and effectively absorbs X-rays.
It is therefore used as a contrast agent in
X-rays of the digestive tract (§3.2,
section "Contrast agents") and as a component of barite
plasters, which cover the walls of X-ray examination
rooms to prevent unwanted radiation into the surrounding areas.
Natural barium is a mixture of 7 stable
isotopes: 130Ba (0.11%), 132Ba (0.1%), 134Ba (2.42%), 135Ba (6.59%), 136Ba (7.85 %), 137Ba (11.23%), 138Ba (71.7%). Of the radioactive isotopes of barium, only
133-Ba has practical use :
Barium 133 Ba
With a half-life of 10.54 years, is
converted by electron capture to excited states 133Cs; during its
deexcitation, photons of g-radiation
are emitted mainly with energies of 81, 276, 303, 356 and 384
keV. After electron capture, photons of characteristic X-rays
of cesium Ka 30.6-31 keV (60%) and Kb 34.9-36.8 keV
(18%) and Auger electrons are further emitted. The 133Ba isotope is
prepared by proton irradiation in a cyclotron by the reaction 133Cs (p, n) 133Ba.
For the proximity of the main energy gamma
356keV 133-barium to the energy 364keV 131I, barium pointers are also used in
some workplaces of nuclear medicine to mark the position and
structures in thyroid scintigraphy with radioiodine (§4.9.1 "Thyrological radioisotope
diagnostics"). Sometimes 133Ba is also used as a calibration standard for
radiometers.
Note: In the past, the isotope 133-Ba
has been tested as a tracer radioindicator to determine radium
kinetics in the environment.
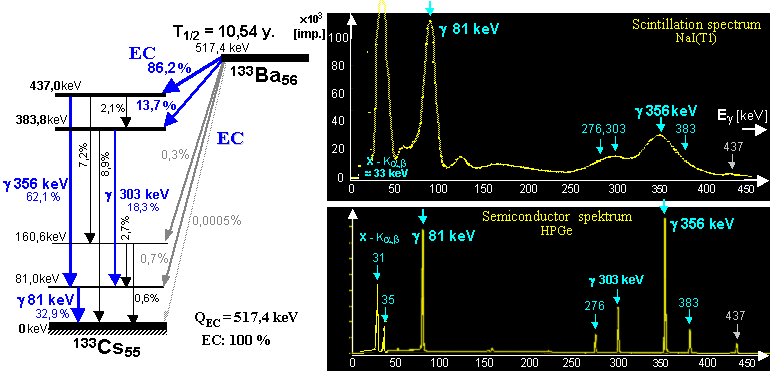
Conversion scheme and gamma spectrum of barium 133Ba.
Samarium
Samarium Sm 62 is a soft silvery metal from the lanthanide group (its discoverer P.E.L.Biosbaudran in 1879 this
name derived from mineral samarskit , which y.1847 in
South Ural found V.ESamarskij -Bychovec). In terrestrial nature, it occurs in the form of
compounds, the concentration in the earth's crust is about 6
mg/kg. In combination with cobalt, samarium is used to make
strong permanent magnets. Samarium has 5 stable isotopes: 144Sm (3.07%), 149Sm (13.82%), 150Sm (7.38%), 152Sm (26.75%), 154Sm (22.75%). Natural
samarium also contains two very long- lived radioactive isotopes 147Sm (14.99%) and 148Sm (11.24%) :
Samarium 147 Sm
with a half-life of 1.06.1011 years is transformed by alpha-radioactivity
(Ea =
2.31MeV) to a basic state of 143Nb. This primordial radioisotope 147Sm with its daughter product 143Nb is used in nuclear geochrology (see "Radioisotope
(radiometric) dating"
above).
Samarium 148 Sm
with a half-life of 7.1015 years is converted by alpha-radioactivity
(E a =
1.96MeV) to a baseline of 144Nb. This primordial radioisotope has no use.
The isotope samarium-153 is artificially
produced :
Samarium
153 Sm
is converted with a half-life of 46.5 hours by beta- -radioactivity (Ebmax = 808keV) to the stable isotope 153Eu. Conversions take place in 18.4% to the ground state
(Ebmax = 803keV) and further to excited
states 153Eu
103keV (49.4%) and 173keV (31.3%). Only in a small percentage
(<0.02%) higher excited states of 270, 585, ...., 764 keV
occur. With the gradual deexcitation of 153Eu, a larger number of transitions take place with the
emission of gamma-photons with energies mainly 69.7keV (4.7%) and
103keV (29%). Conversion electrons are emitted by the internal
conversion of excited levels and a characteristic X-ray with an
energy of 40-48 keV is generated.
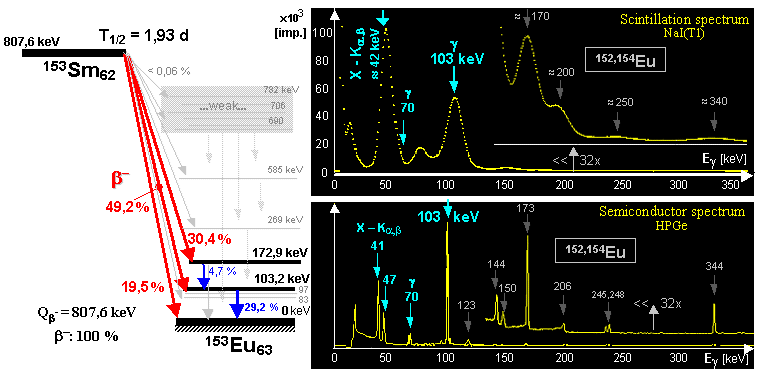
Conversion scheme and gamma spectrum of
samarium 153Sm.
Note: In the
right part of the spectrum, some gamma peaks from radionuclide
impurities 152Eu and 154Eu are shown in an enlarged section (a detailed spectrum
of radionuclide contaminants is shown in the figure below).
In the gamma spectrum of 153 Sm, in addition to braking radiation, the distinctive
70keV and 103keV photopeaks are evident; peaks of higher energies
are visible only at higher magnification. At the beginning of the
spectrum, there is a characteristic X-ray with lines Ka,b with
an energy of about 45 keV, arising during the internal conversion
from excited levels of 172 and 103 keV.
Radionuclide impurities in samarium-153
In our spectrum of the sample samaria-153 ("
Quadramet ") no weak higher
gamma peaks are evident, as they are over-radiated with radionuclide
Europium contaminants 152Eu and 154Eu. The image below shows the gamma
spectrum of the long-term contaminants themselves, which was
measured with a 3-month-old samarium sample, in which the
samarium-153 itself has already completely decomposed, leaving
only impurities with longer half-lives :
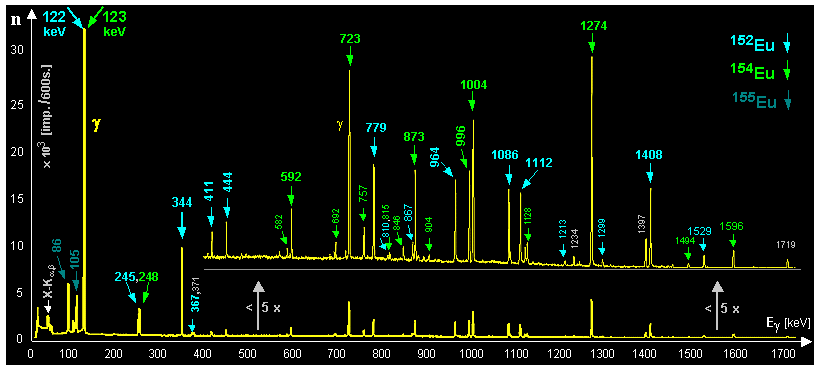
Gamma spectrum of long-term radionuclide impurities in a 3-month
"extinct" samarium sample 153Sm.
In the spectrum containing a large number
of gamma-peaks, we found three radioactive isotopes of europium: 152Eu
(T1/2 =
13.5 years; its physical properties and gamma-spectrum are given
below in the passage Europium 152 Eu ), 154Eu
(T1/2 =
8.6 years) and 155Eu
(T1/2 =
4.7 years). The respective photo peaks of these isotopes are
marked according to the color assignment at the top right.
The 153Sm isotope is
prepared in a nuclear reactor by irradiating samarium (enriched with the isotope 152Sm to 99%) with neutrons by the
reaction 152Sm (n, g) 153Sm. From the trace isotope
impurities in the target, the above-mentioned radionuclide
impurities 152,154,155Eu are also formed by neutron fusion.
Beta-radiation of samarium-153 with the
phosphate ligand EDTMP (which binds to bone
hydroxyapatite at sites of increased osteoblastic activity) is used in nuclear medicine for palliative therapy
of bone metastases of the breast and prostate ca (§3.6, section "Radioisotope
therapy").
Europium
Europium Eu 63 is a soft shiny metal from the lanthanum
group. In terrestrial nature, it occurs only in compounds and is
relatively sparsely represented (approximately 1.2 mg/kg in the
earth's crust). The most common use of europium is in luminescent
materials. Europium has two stable isotopes 151Eu (47.8%) and 153Eu (52.2%) (at 151Eu was recently discovered small a-radioactivity with
an extremely long half-life of about 5.1018 years - in
practical terms, but it can considered stable). Of the radioactive isotopes, europium-152 is somewhat
important :
Europium
152 Eu
With a half-life of 13.54 years, it is
transformed by a very complex branched disintegration scheme. In
27.9% there is b- -radioactivity to excited levels of 152Gd, in 71.9% electron
capture (in a small percentage
also competitive beta+ -conversion) to excited levels of
152 Sm *).
During deexcitation of the excited states of daughter
gadolinium-152 and samaria-152, a large amount (almost 100) of gamma-ray
photons is emitted, of which photons with energy have significant
intensity: 122, 245, 344, 779, 867, 964, 1086, 1112 and 1408 keV.
For this large energy range of distinctive gamma lines, 152Eu is used as a
advantageous calibration standard in gamma-spectrometry
(Chapter 2 "Detection and
spectrometry of ionizing radiation",
§2.4, section "Gamma-ray spectrometry").
*) The daughter samarium 152Sm is stable,
but the daughter gadolinium 152Gd is slightly radioactive - with a
huge long half-life of 1.1.1014 years, it is
converted by alpha-radioactivity to 148Sm (and then with
even longer half-lives by two more alpha-conversions to 144Nd and finally to
stable 140Ce).
The radiation of these tertiary radionuclides is completely
negligible.
The 152Eu isotope is formed
by irradiating europium with slow neutrons in a nuclear reactor
in a 151Eu
(n, g) 152Eu reaction.
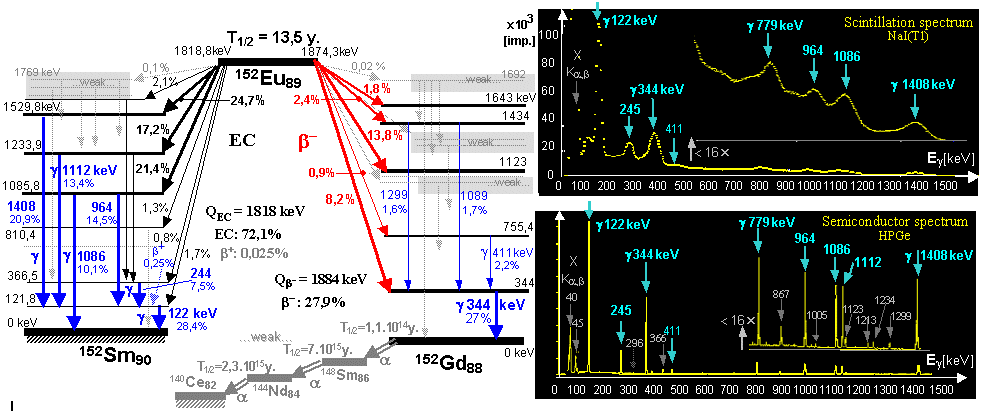
Disintegration scheme and gamma spectrum of europium 152Eu
Europium 152m Eu
is one of the few metastable nuclei (isomers) that have such different
quantum properties from the ground state (especially the
spin value) that there is no usual transition to
the ground state of g- radiation photon emissions, but to the radioactive
conversion of beta-, beta+ or electron capture, to another
adjacent nucleus (§1.2, passage "Nuclear isomerism and metastability"). 152mEu does not de-excite at
all to basic 152Eu, but with a
half-life of 9.31 hours it is converted from 73% by b--radioactivity
to 152Gd and from 27% by electron capture and b+
to 152Sm. The 152mEu isotope is formed by
irradiating europium in a nuclear reactor by reaction 151 Eu (n, g ) 152m Eu.
The relatively
"exotic" isotope 152mEu has no practical use
and in addition to the special non-photon conversion of the
isomer, we mention it here also because it was used in 1958 by
M.Goldhaber et al. to determine the so-called helicity of a
neutrino (§1.2, part "Neutrinos - "ghosts"
between particles",
passage "Difference between neutrinos and
antineutrinos - helicity of neutrinos. Goldhaber's experiment.").
Europium 154 Eu
with a half-life of 8.6 years from 99.8 %
is converted by beta-
-radioactivity to excited levels of 154Gd, from 0.2 % by electron capture to excited
states of 154Sm.
During deexcitation, gamma radiation is emitted mainly with
energies of 123, 248, 592, 723, 996, 1005 keV.
Europium 155 Eu
With a half-life of 4.7 years, is converted
to excited levels and to a baseline state of 155Gd by beta-
-radioactivity. During deexcitation,
gamma radiation is emitted mainly with energies of 87 and 105
keV.
Mixed gamma spectrum of europium isotopes 152,154,155Eu can be seen above in the spectrometric measurement of
radionuclide impurities in the
samarium-153 sample.
Gadolinium
Gadolinium Gd 64 is a ductile shiny metal from the group of lanthaniodes.
In terrestrial nature, it occurs only in compounds and is
relatively sparsely represented (approximately 6.2 mg/kg in the
earth's crust), it is mainly mined from the mineral monazite.
Gadolinium has interesting magnetic properties: at lower
temperatures below 20 °C it is ferromagnetic, at higher
temperatures paramagnetic. It is used as a contrast agent
in nuclear magnetic resonance MRI. Another use of gadolinium is
in luminescent materials in radiology. The 157Gd isotope has a
very high effective cross section (approximately
260,000 barn) for thermal neutron capture,
so it is used in some nuclear reactors.
Gadolinium has six stable isotopes: 154Gd (2.2%), 155Gd (4.8%), 156Gd (20.5%), 157Gd (15.6%), 158Gd (24.8%) and 160Gd (21.9%). Natural
gadolinium also contains a natural radionuclide of primordial
origin 152Gd
(0.2%) with a very long half-life of about 1014 years (alpha radioactivity is converted to 148Sm).
Of the radioactive isotopes,
gadolinium-153 is occasionally used :
Gadolinium
153 Gd
With a half-life of 240.2 days, it is converted
by electron capture to 153Eu in the ground state (4%) and in the excited states of
97keV (38%), 103keV (42%) and 173keV (16%). During dexcitation, gamma
photons are emitted mainly with energies of 97 and 103 keV. At
the beginning of the spectrum there are significant peaks Ka,b of
the characteristic X-ray of europium with
energies around 44keV.
The 153Gd isotope is mostly prepared from europium. Irradiation
with neutrons in a nuclear reactor produces a combination of: 153Eu(n,g)152mEu(b-,T1/2=92min)®153Eu® 152Gd; 152Gd(n,g)153Gd. Or irradiation with alpha-particles in a cyclotron
produces a sequence of processes: 151Eu(a,2n)153Tb(b+,T1/2=2,3d)®153Gd.
Gadolinium 153Gd was used as a
two-photon source of gamma radiation 103keV and X 44keV in bone
densitometry by the DEXA method (§3.2,
passage "Bone densitometry"), experiments were also performed using CT. Sometimes they are used gadolinium
rods in SPECT scintigraphy for correction
of gamma radiation intensity on attenuation during tissue passage
(§4.3, passage "Adverse
effects of SPECT and their correction",
point "Absorption of gamma radiation"). However, these methods have not worked very well and
are mostly already abandoned ...
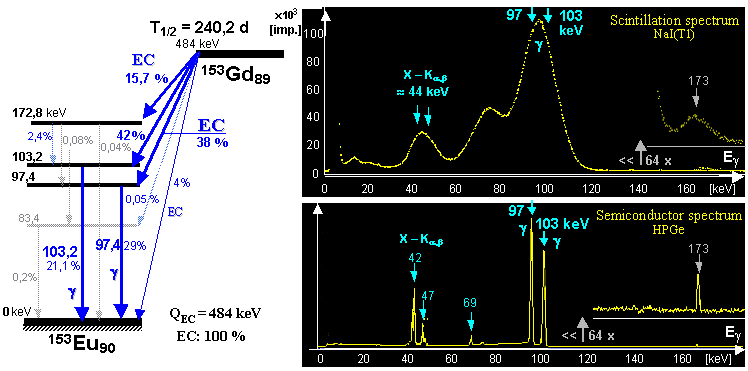
Conversion scheme and gamma spectrum of gadolinium 153Gd.
Erbium, Yterbium, Terbium
The names for some lanthanides "terbium, yttrium,
erbium, yterbium" originated historically from the
Swedish village of Ytterby, near which these elements
were first discovered in a feldspar quarry in the minerals
gadolinite and monazite.
Erbium Er
71 is a soft silvery shiny metal from the lanthanide
group. It is contained in the earth's crust in the form of
compounds in a concentration of about 2.5 mg/kg. It has 6 stable
isotopes: 162Er (0.14%), 164Er (1.6%), 166Er (33.5%), 167Er (22.87%), 168Er (26.98%), 170Er (14.91%). Of the radioactive isotopes, one has
practical significance :
Erbium
169 Er
is an artificially produced radionuclide, which with a half-life
of 9.39 days is converted by b--radioactivity from 55% to basic (Eb max = 353keV) and from 45% to a low excited state of 169Tm with energy
8.41keV (at deexcitation emits low-energy
radiation g of this energy). With a very low
intensity, an excited state of 118keV arises. Erbium-169 is a practically
pure beta emitter, as soft gamma radiation of 8.4 kV is
subject to almost 99% internal conversion, especially on the M
and N shells of the Tm atom (with
significant emission of low-energy conversion electrons 0.7-8.4
keV, approx. 250 %) and line 110, 118keV is
primarily very weak.
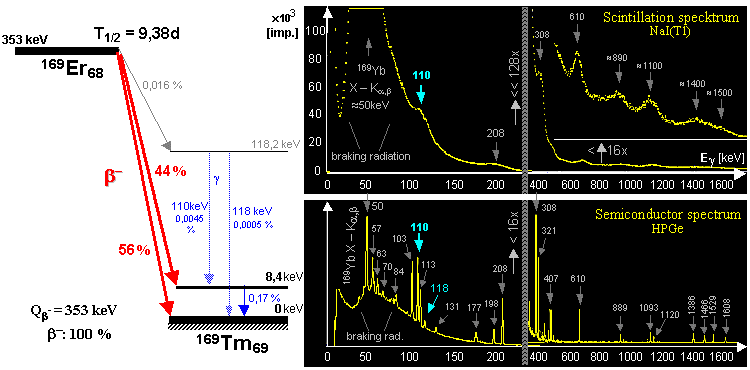
Conversion scheme and gamma spectrum of
erbium 169Er.
Note: Erbium 169 has only two
very weak gamma peaks 110 and 118
keV (indicated by blue arrows in the spectrum). A number of other
peaks measured in our spectrum come from small radionuclide
impurities that the measured sample of erbium-169
contained. We mainly measured yterbium 169Yb with peaks: 50.57.59keV (XK a, b Tm), 63, 110,
118, 130, 177, 198, 308 keV (its physical properties and gamma
spectrum are given below in the passage Yterbium
169 Yb ). Further samarium 153Sm: 70, 103 keV (its physical
properties and gamma spectrum are given above in the passage Samarium
153 Sm ); erbium 172Er
: 407, 610 keV; lutetium 177Lu : 113, 208, 250, 321 keV (its
physical properties and gamma spectrum are given below in Lutetium
177 Lu ); finally trace
amount of thulium 172Tm : 79, 182, 1093, 1120, 1387, 1466,
1529, 1608 keV. Due to the very weak gamma radiation of the own 169Er, even a small
amount of these impurities (<0.1%) is significantly reflected
in the spectrum, even the own gamma erbia-169 overlaps.
After all, peaks 110 and 118 keV belonging to 169Er also has a
contaminant of 169 Yb, so in our sample they may not have even come from
erbium-169 ..?..
The gamma spectrum of 169 Er consists mainly
of relatively soft braking radiation with a continuous spectrum,
which with a decreasing tendency stretches up to about 300keV.
With longer acquisition, barely visible gamma peaks of 110 and
118 keV appear on this continuous background.
From a spectrometric
point of view, the radionuclide 169Er itself is quite
uninteresting and dull - only two very faint, barely
visible gamma peaks of 110 and 118
keV can be seen against the continuous background of the braking
radiation. However, what was our surprise when we had a sample of
about 100kBq of erbium-169 preparation (citrate
intended for radiation synovectomy of small joints) inserted into a sensitive gamma-spectrometer: after
several tens of minutes of acquisition, the spectrum was
literally "bristling" by a number of gamma-peaks,
which, of course, did not belong to 169Er, but to radionuclide impurities
*) - see the picture above with accompanying text.
*) From the spectrum in the picture, it
might seem at first glance that there are unbearably many of
these radionuclide impurities..?.. However, it is just an
"optical illusion"! Erbium-169 has a very low
proportion of gamma radiation - only about every 20,000
transformations, one detectable 110keV gamma photon is emitted.
However, those impurities such as ytterbium-169, samarium-153,
...., have many times higher gamma emissions (about 1000 times),
so even their very small concentration (at the level of hundredth
of %) will be more pronounced in the spectrum than alone 169Er ..!..
The isotope 169Er is prepared in a
nuclear reactor by neutron irradiation of erbium oxide (enriched in the isotope 168Er) by the reaction 168Er (n, g) 169Er.
Erbium-169 is used in the form of a
colloidal solution of citrate in nuclear medicine for radiation synovectomy
of small joints (fingers, wrists) - due to the low energy of
emitted beta-particles, with a range in the tissue of about 1 mm.
Yterbium
Yb 70 is also a soft silvery white metal from the lanthanide
group. It is contained in the earth's crust in the form of
compounds in a concentration of about 2.5-3 mg/kg. It has 7
stable isotopes: 168Yb (0.13%), 170Yb (3.04%), 171Yb (14.28%), 172Yb (21.83%), 173Yb (16.13%), 174Yb (31.83%), 176Yb (12.76%).
Occasionally, one artificial radioisotope
of ytterbium is used :
Ytterbium
169 Yb
with a half-life of 32.02 days converted by electron
capture (EC), into a number of excited states of thulium
169Tm:
316keV (5%), 379keV (82%), 473keV (13%); conversions
to other higher excited states have a very low intensity
(<0.01%). In cascade deexcitation, many
energies of gamma photons are emitted, of which the energies of
63, 110, 130, 177, 198 and 308 keV have a more significant
intensity.
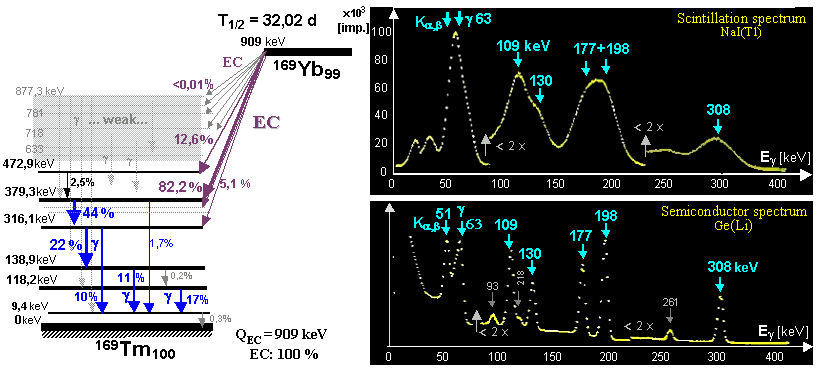
Conversion scheme and gamma spectrum of ytterbium 169Yb. Apology: The semiconductor
spectrum was measured on an older Ge(Li) detector with degraded
properties.
The gamma spectrum of 169Yb is dominated by
the peaks of the characteristic X-ray Ka,b 49-59 kV,
followed by the gamma peaks 63, 110, 130, 177, 198 and 308 keV.
The peaks from the 139-->118keV and 9.4-->0keV transitions
are weak because more than 99% are subject to internal
conversion. A number of other higher peaks in the range of
316-781keV are so low (<0.01%) that in our weak sample (approx. 3kBq) they did not
displayed in the spectrum.
The isotope 169Yb is prepared in a cyclotron by irradiating thulia
oxide with protons in a 169Tm (p, n) 169Yb reaction . ... and other
reactions .....
Yterbium-169 has been used in nuclear
medicine for scintigraphic diagnostics,
especially of the cerebrospinal fluid
(§4.9.8, passage "Scintigraphy of
cereprospinal fluid pathways "). 169Yb is also being investigated as a potential
radioisotope for brachytherapy (§3.6, section "Brachyradiotherapy") or defectoscopy
(§3.3, section "Radiation defectoscopy") with a lower average gamma
energy (93keV) than the commonly used iridium 192 Ir.
Terbium Tb
65 is another soft silvery shiny metal element from the
lanthanide group. It is contained in the earth's crust in the
form of compounds in a concentration of about 0.9 mg/kg. It was
used in phosphors in screens and in X-ray intensifying films -
has been abandoned, replaced by electronic digital imaging; it is
now used as part of the recording layers of magneto-optical
media.
Terbium has one stable isotope of 159Tb (100%). Of the
radioactive isotopes of terbium, the four radionuclides are being
tested experimentally in nuclear medicine :
Terbium
149Tb
This artificially prepared radionuclide is transformed with a
half-life by a very complex branched decay
scheme with a half-life of 4.12 hours. In 76.2% electron
capture occurs and in 7.1% b+
-radioactivity takes place (mean Eb + = 730keV), in
both cases to excited levels of 149Gd. In 16.7%, alpha-radioactivity (Ea =
3.97MeV) reaches a baseline state of 145Eu. During deexcitation of the excited states of
daughter gadolinium-149, a large amount (more than 300!) of gamma-ray
photons is emitted, of which photons with energy
have significant intensity: 165keV (26%), 352keV (29%), 389keV
(18%), 652keV (16%), 817keV (12%), 853keV (15%); high energy
gamma peaks up to more than 3000keV are weak.
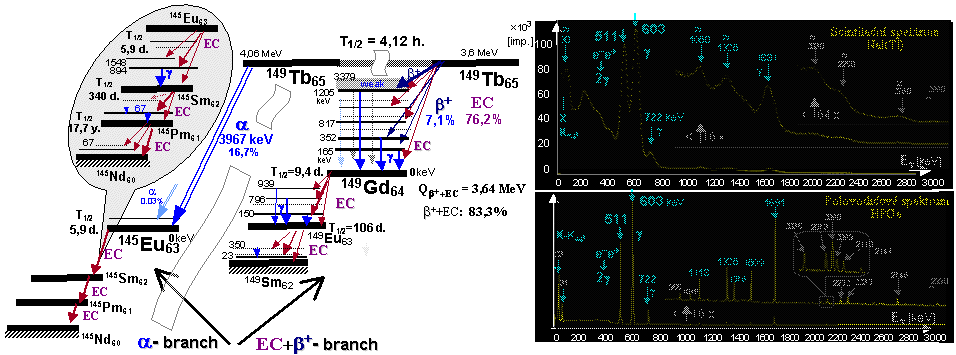
Conversion scheme and gamma spectrum of
terbium 149Tb ( .. The gamma-spectrum
comes to be measured when I manage to obtain a sample of 149Tb
...) .
Due to lack of space, the complex decay
scheme of 149Tb is drawn in a simplified way, without
details of weakly represented energy levels and transitions.
Both daughter radionuclides are again
radioactive. The daughter gadolinium 149Gd
from the e - b+ branch
is transformed by electron capture to a number of
excited states of europium 149Eu with energies mainly 150, 490, 790, 950 and 1083 keV,
with subsequent emission of a number of gamma radiation lines.
This europium 149Eu is then again converted by electron capture
by a half-life of 106 days into excited states of the already
stable samarium 149Sm. Daughter europium 145Eu from alpha -branches with a half-life of
5.9 days are first converted by electron capture to excited
states of samarium 145Sm with emissions of a number of gamma lines, mainly
with energies of 653, 893, and 1658 keV. This samarium is then
again converted to 145Pm with a half-life of 340 days by electron capture,
which with a half-life of 17.7 years is finally converted to a
stable neodymium of 145Nd also by e-capture.
Preparation : 149Tb is still a
non-standard isotope and in small quantities it is experimentally
prepared in three ways 1. Direct nuclear
reactions with accelerated protons (energy
approx. 100MeV) or helium nuclei: 149Gd (p, 4n) 149Tb, or 151,153Eu (3, 4 He, x n) 149Tb; 2. Reactions induced by accelerated
heavier ions, eg nuclei 12C: 141Pr(12C, 4n)149Tb, or 142Nd(12C, 5n)149Dy (b+,EC, T1/2 4min.)®149Tb; 3. Fragmentation reactions of
high-energy protons, eg on a tantalum target: Ta (p, ..x ..) 149Tb, with isotope
separation.
For nuclear medicine, 149Tb terbium is
interesting that it emits both alpha particles
and beta+ positrons.
In the chelating combination of 149Tb with suitable target biomolecules, the same
preparation (radiopharmaceutical) can be used both for diagnostic
imaging of tumor lesions by positron emission tomography PET *)
and for subsequent biologically targeted radionuclide therapy
with use alpha-particles, with possible simultaneous PET monitoring
of the course of therapy - ie for teragnostics
(§4.9, passage "Combination of diagnostics and therapy -
teragnostics"); it is a "monoteranostic"
radionuclide.
*) For gamma imaging, it is alternatively possible to use
single-photon planar or SPECT scintigraphy using gamma radiation
165 or 352 keV, emitted during deexcitation of the daughter 149 Gd; due to the
large amount of spurious high- g radiation, however, image
quality is not good.
The 149Tb radionuclide has
so far been experimentally tested in radioimmunotherapy of
lymphoma for the designation of rituximab using DOTA or
DTPA chelators, with preliminary useable scintigraphic and
therapeutic results.
The problem of the teranostic use of 149Tb can be a complex
decay scheme, containing several daughter radioisotopes,
some long - lived. Due to the relatively short half-life of 4.1
hours is 149Tb suitable for labeling lower molecular weight
radiopharmaceuticals that are rapidly removed from the body. In
this case, the potential radiotoxicity daughter radiolanthanides (converting the low load electron capture) did not significantly manifest. Due of its physical
properties, terbium-149 would remain only as a curiosity, it will
probably not work as teranostic
radionuclide..?..
Terbium 152Tb
with a half-life of 17.5 hours, is transformed by electron
capture (79.7%) and beta+-radioactivity
(20.3%) into excited states of 152Gd. During deexcitation, gamma photons with energies of
271 keV (9.5%), 344 (63.5%), 686 (3.2%), 779keV (5.5%) are
emitted, and during positron annihilation, gamma photons of 511
keV (41% ).
Terbium-152 can be used in nuclear medicine for PET scintigraphy.
Rather than for basic PET diagnostics, it is tested in a theranostic
approach, as an accompanying diagnostic isotope in
PET/CT monitoring of the biodistribution of therapeutic ligands
labeled with terbium l61Tb - the theranostic pair ll52Tb/l61Tb.
Terbium 155Tb
with a half-life of 5.32 days is converted by electron
capture (100%) to excited states of 155Gd. During
deexcitation, gamma photons with energies of 105.3 keV (25.1%),
180.1 (7.5%), 262.3 keV (5.3%) are emitted.
Terbium-155 can be used in nuclear medicine for (single-photon)
planar/SPECT scintigraphic imaging, in principle similar to
iodine 123I or indium 111In. However, its main
potential use is expected in the theranostic
approach, as a diagnostic isotope in scintigraphic monitoring of
the biodistribution of therapeutic ligands labeled with terbium l61Tb - the theranostic
pair l55Tb/l61Tb.
Terbium 161Tb
is an artificially produced radionuclide, which with a half-life
of 6.9 days is converted by beta--radioactivity (Ebmax=593keV) to 161Dy - from 10% to the ground state, further to the
excited states 25.6keV (10%), 74.5keV (65%), 131,7keV (25,7%),
and other five higher energy levels in the range of 213-550 keV
with a very low proportion of about 0.01-0.06. During
deexcitation of excited 161-dysprosium nuclei, gamma
radiation photons are emitted with energies mainly
25.6keV (23%), 49keV (17%), 74.5keV (10%), 88keV (0.2%), with low
intensity (approx. 0.001-0.1%) then higher energy in the range of
100-550 keV. At the beginning of the spectrum are the peaks of
the characteristic X-ray (Ka,b Dy) 45-54 keV (0.3-12%).
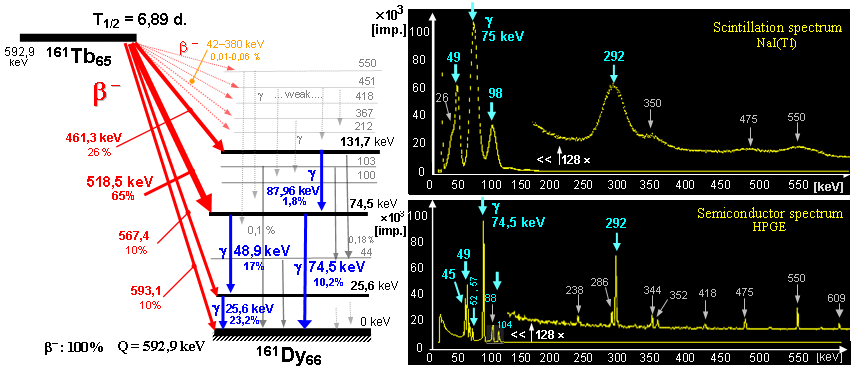
Conversion scheme and gamma-spectrum of terbium 161Tb .
The 161Tb sample was prepared at the reactor at ÚJV
Rež on the initiative of RNDr. Martin Vlk, PhD. The
semiconductor spectrum was then measured with the cooperation of
Ing. Matej Grapa in the SÚRO Spectrometric Laboratory in
Ostrava. The author is very grateful to both colleagues.
The radionuclide 161Tb is usually
produced in a nuclear reactor by neutron irradiation of
gadolinium highly enriched in the 160Gd isotope, using the reaction 160Gd(n,g)160Gd(b-, T1/2 3,6min.) -->161Tb.
Terbium-161 is currently used
experimentally in nuclear medicine for biologically targeted radioisotope
therapy of cancer (§3.6, section
"Radioisotope therapy"). 161Tb has similar properties and therapeutic uses to the
already successfully used lutetium 177Lu , compared
to which a slightly increased therapeutic efficacy is expected,
due to the greater proportion of conversion and Auger electrons/1 decay. A 161Tb-DOTA chelate conjugate with an octreotide
peptide, which binds to neuroendocrine tumor cells with a
sufficiently high concentration of somatostatin receptors,
can be used to treat neuroendocrine tumors. However, 161Tb-labeled monoclonal
antibodies, such as 161Tb-PSMA617 anti-prostate specific membrane
antigen (PSMA), show very good results.
These methods are
briefly described in §3.6, section "Radioimunoterapy".
Beta radiation with
a mean energy of 154 keV (100%) and also conversion and Auger
electrons (which are more abundant in one
decay than in 177Lu) have a therapeutic effect.
Emission of gamma radiation of 74.6 keV
enables simultaneous scintigraphic imaging
of tissues and organs to which the therapeutic preparation has
penetrated. On these scintigraphic images, we can assess (visually, or even quantitatively)
the degree of desirable uptake of radiopharmaceuticals in tumor
foci and unwanted accumulation in healthy tissues and critical
organs - to monitor the course of radioisotope
therapy. The representation of gamma radiation also
allows easy measurement of the activity of 161Tb preparations in
conventional activity meters with the ionization chamber (§2.3 "Ionization chambers"), using the appropriate
gamma-coefficient calibration.
An excellent teranostic pair
of 155Tb for planar/SPECT
scintigraphic diagnostics, or 152Tb for PET, and 161Tb for biologically targeted radionuclide therapy, is also
being considered (mentioned above). These are radionuclides of
the same element Tb, with identical
chemical and therefore biochemical properties and metabolism.
Lutetium
Lutecium Lu 71 is a soft white shiny metal, the last member of the
lanthanide group, which in the form of compounds is relatively
rare in nature (0.5-0.7 mg/kg in the earth's crust). It is used
in LSO scintillation detectors for positron
emission tomography (§2.4., part
"Scintillators and
their properties" and §4.3,
part "PET cameras"). It has the only stable isotope 175Lu (97.4%), while natural lutetium also contains 2.6% of
the isotope 176Lu, which is radioactive :
Lutetium
176 Lu
is a natural (primordial) radionuclide which, with a very long
half-life of 3.76.1010 years b- -radioactivity, is converted to stable
hafnium 176Hf
- to an excited state of 597 keV. Deexcitation occurs by gradual
(cascade) emission of gamma photons with energies of 307, 202 and
88keV (a higher energy of 401keV is weakly
represented, a weak summation peak 306+202 keV also appears in
the spectrum). It is used for long-term
dating in nuclear geochronology using the lutetium-hafnium
method (see "Radioisotope dating" above). The content of
natural 176Lu
has a somewhat adverse effect on "internal
contamination" LSO
scintillators and their relatively high
radiation background (see §2.4
"Scintillators and
their properties" and §4.3
"PET cameras").
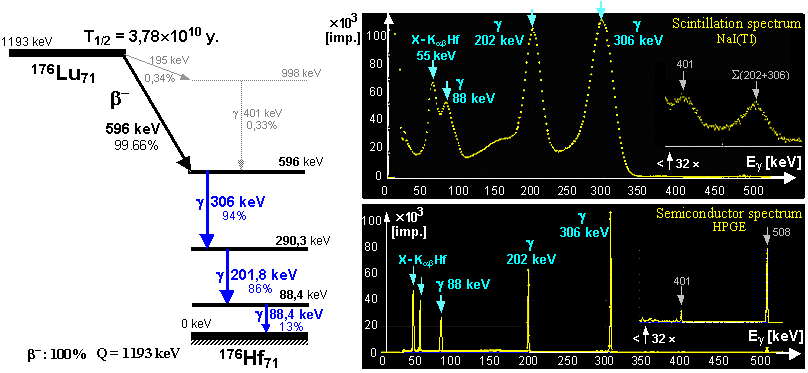
Conversion scheme and gamma spectrum of lutetium 176Lu
Lutetium
177 Lu
is an artificially produced radionuclide, which with a half-life
of 6.65 days is converted by beta- -radioactivity (Eb max = 498keV) to 177Hf - from 79.4% to the ground state, further to excited
states 113keV (9%) and 321keV (11.6%). During the deexcitation of
excited 177-hafnium nuclei, photons of gamma radiation
with energies mainly 113keV (6.2%) and 208keV (10.4%) are
emitted, with low intensity (approx. 0.2%) then 72, 250 and 321
keV. At the beginning of the spectrum, the peak of characteristic
X-radiation (Ka,b Hf)
55-63 keV is visible.
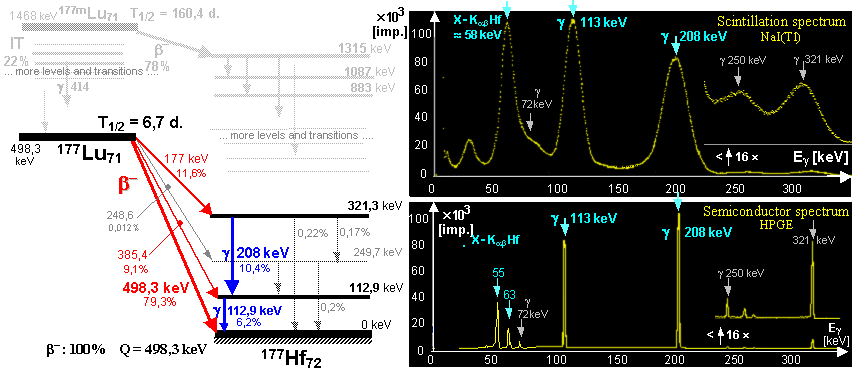
Conversion scheme and gamma spectrum of lutetium 177Lu.
For the sake of interest, a simplified decay scheme of the
metastable isomer 177mLu, which often forms together with 177Lu, is also drawn in
the upper left left .
The 177Lu isotope is
produced by irradiating neutrons in a nuclear reactor in two ways
:
- The direct method
consists in irradiating lutetium enriched in the 176Lu isotope with
neutrons in the 176Lu (n, g) 177Lu reaction. Due to the high effective cross section of
this reaction, a radionuclide with a relatively high specific
activity can be obtained. A certain disadvantage of this process
is the ongoing reaction of 176Lu (n, g) 177mLu, which produces the nuclear isomer 177mLu with a half-life
of 160 days. This unwelcome contaminant is dexcited in 22% by
gradual isomeric transitions to the ground state of 177Lu and in 88% is
converted separately b--radioactivity
(Eb max = 153keV) to a
relatively high excited state of 1315keV of the daughter 177Hf, which deexcites
through a series of g- transitions towards lower levels. At this radioactivity,
177mLu
emits a number of gamma energies, some of which are the same as
for 177Lu,
but higher energies of 228, 281, 319, 378, 418 keV and many
others are also represented. Contaminant radiation 177mLu increases the
radiation exposure for a long time and deteriorates the quality
of scintigraphic images.
- Indirect method consisting in the combination
of neutron irradiation of ytterbium, enriched in the isotope 176Yb, neutrons in the
reaction 176Yb (n,g) 177Yb and subsequent beta-conversions of 177Yb (b-, T1/2 = 1.9
hours) ® 177Lu to the resulting
lutetium-177. This method does not produce interfering 177mLu (with the beta-conversion of 177Yb, the metastable level of 970keV 177mLu is not saturated), only trace
amounts of Yb isotopes may be present....
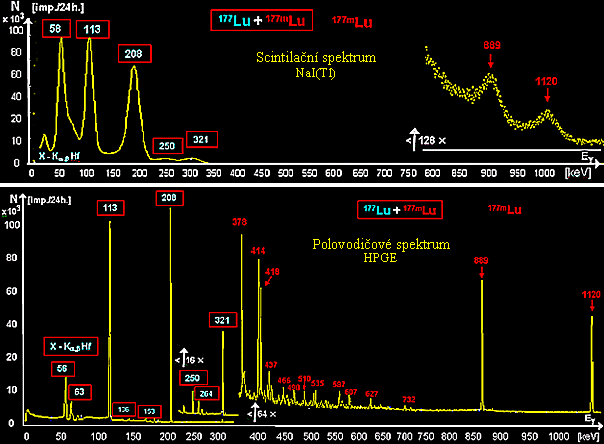 |
Gamma spectrum of a 3 month old sample
177Lu (Betalutin preparation) .
Apology: During the graphic processing of
the spectra, the scales on the horizontal axis were
deformed. However, the numerical values of the energies
above the peaks are accurate.
|
Lutetium-177 is used in nuclear medicine for
biologically targeted radioisotope therapy of
tumors (§3.6, section "Radioisotope therapy"). There is marginal use in
the form of 177Lu-EDTMP for the palliative therapy of bone
metastases (since strontium 89 Sr, samaruim 153 Sm and rhenium 186 Re have been proven
for this purpose). More common is the
therapy of neuroendocrine tumors using a
conjugate of chelate 177Lu-DOTA with the peptide octreotide,
which binds to neuroendocrine tumor cells with a sufficiently
high concentration somatostatin receptors. However, very
good results are shown in particular by 177Lu- labeled monoclonal antibodies,
such as 177Lu-J591 anti-prostate specific membrane antigen
(PSMA) or 177Lu-tetulomab anti-CD-37 for the treatment of
lymphomas.
These methods are described in §3.6,
section "Radioimmunotherapy".
Beta
radiation with a mean energy of 134keV has
a therapeutic effect. The advantage of 177Lu is that gamma emission
allows simultaneous scintigraphic imaging
in therapy tissues and organs into which the therapeutic
preparation has penetrated. On these scintigraphic images we can
assess (visually, or even quantitatively) the degree of desirable uptake of radiopharmaceuticals
in tumor foci and unwanted accumulation in healthy tissues and
critical organs - to monitoring the course of
radioisotope therapy. The representation of gamma
radiation also allows easy measurement of the activity
of 177Lu
preparations in conventional ionization chamber activity meters (§2.3 "Ionization
chambers"), using the appropriate gamma-constant calibration.
Tungsten, Rhenium
Tungsten (wolfram) W 74 is
a gray-white shiny, heavy (density 19.25 g/cm3) and
difficult-to-melt metal (melting point 3422 °C). It is
relatively rare in nature (approx. 1.5-3.5 mg/kg in the earth's
crust), in compounds (ores - oxides) together with iron, manganese, calcium or lead. Tungsten
has a wide technical application due to its high density and heat
resistance. For high melting points, it is used as a material for
filaments in light bulbs and tube cathodes
(including electron sources in accelerators). Tungsten alloys are
used in metallurgy to increase the hardness,
mechanical and thermal resistance of metals. Due to its high
density, it is used as effective radiation shielding
of X-ray and gamma radiation (§1.6,
section "Radiation absorption
in substances", section "Gamma
radiation shielding"),
for the production of containers and precision collimators.
It has 5 stable isotopes: 180W (0.12%), 182W (26.5%), 183W (14.31%), 184W (30.64%), 186W (28.43%).
Note: The 180W isotope was found
to have very weak radioactivity - alpha-conversion
to hafnium 176Hf with a hugely long half-life of 1.8.1018 years. It is
suspected that other natural isotopes of tungsten are perhaps in
the long run a-radioactive (converted to the corresponding hafnium
isotopes), with even longer half-lives. It is very difficult to
verify (in a sample of 1 gram of tungsten
it would represent less than one alpha-conversion per year).
Of the radioactive
isotopes of tungsten, only one is worth mentioning here :
Tungsten 188 W
With a half-life of 69.4 days, is converted by b-
-radioactivity from 99% to the ground state of rhenium 188Re, from 1% to
several excited levels, during which deexcitation emits several
photons of gamma radiation, especially with energies of 227 and
290 keV. The 188W isotope is prepared by neutron irradiation of metallic
tungsten (enriched to about 95% with the isotope 186W) in a nuclear
reactor by double neutron capture: 186W (n, g) 187W (n, g) 188W. It serves as the parent isotope in the 188W/188Re
generator to obtain the 188Re radionuclide in nuclear medicine (see Re-188 below).
Rhenium Re 75 is also a very heavy, hard and difficult-to-melt metal,
very rarely present in nature (approximately
1-5 ng/kg in the earth's crust). Natural
rhenium is a mixture of 37.4% of the stable isotope 185Re and 62.6% of the
radioactive isotope 187Re (b-,
half-life 4.33.1010 years).
Three radioactive isotopes of rhenium are of practical importance
:
Rhenium 187 Re
is a natural (primordial) radionuclide, which with a very long
half-life of 4.33.1010 years by b- -radioactivity is converted to the
basic state of osmium 187Os. It is used in long-term radiosotope dating - nuclear
geochronology - for determining the age of rocks by the 187Re/187Os method (see "Radiometric dating" above).
Rhenium
186 Re
is an artificially produced radionuclide which, with a half-life
of 3.72 days, is branched transform in 92.53% by beta- -radioactivity to 186Os and in 7.47% by electron
capture to 186W. b-
transformations take place in 71% to the ground state (Eb max = 1070keV) and in 21.5% to excited levels of 186Os with energy
137keV (in a small fraction of a percentage
also to higher excited levels). Electron
capture takes place in 5.8% to the ground state and in 1.7% to
the excited state 122keV 186W. During deexcitation g photons are emitted
radiation mainly with energy 137keV (9.4%) and 122keV (0.6%).
The isotope 186Re is obtained by neutron irradiation of rhenium (preferably enriched in the isotope 185Re) in a nuclear reactor by the reaction 185Re (n, g) 186Re, or in a
cyclotron by proton irradiation of tungsten (highly
enriched in the isotope 186W) in a reaction of 186W (p, n) 186Re.
Rhenium-186 is used in nuclear
medicine in the form of a phosphonate complex for the
palliative therapy of bone metastases (§3.6, part "Radioisotope
therapy", passage "Radionuclide
therapy of tumors and metastases") and in the form of colloidal sulphite for radiation
synovectomy in rheumatoid arthritis of medium - sized
joints (wrist, elbow, shoulder) - §3.6,
passage"Radionuclide synovectomy".
Decomposition scheme and gamma spectrum of rhenium 186Re.
Rhenium 188 Re
is also an artificially produced radionuclide, which with a
half-life of 16.98 hours is converted by b-
-radioactivity (Eb
max
= 2115keV) to 188Os - from 70.6% to the ground state, from 26% to an
excited level of 155keV, during the subsequent deexcitation,
gamma radiation of the same energy is emitted. With low
representation, beta-transformations also take place in other
excited states of 188Os, whose radiation has a very low intensity.
The 188Re isotope is most often obtained by elution from a 188W/188Re
generator (mentioned above), or is prepared by neutron irradiation of rhenium
(highly enriched in the isotope 187Re) in a nuclear reactor: 187Re (n, g) 188Re. It has been used in recent years in nuclear medicine
for similar applications to the above-mentioned more common 186Re (advantage of 188Re is easier preparation from the generator, higher
specific activity, shorter half-life and higher energy of beta
particles).
Iridium
The heavy noble metal iridium Ir 77 is
rarely present in nature (approximately 0.001 mg/kg in the
earth's crust) with its two stable isotopes 191Ir (29.5%) and 193Ir (62.7%). It has a high density (22.6 g/cm3) and is very
chemically resistant *). Along with platinum, it is used in
metallurgy and a number of technical applications.
*) Known standards of meters and
kilograms were made of an alloy of platinum and iridium
at the International Institute of Weights and Measures
in Sevres, near Paris.
Iridium
192 Ir
A heavy radionuclide often used as
a gamma emitter is iridium 192Ir. With a half-life of T1/2 =
74.2 days, it decays from 95.1% b-
-radioactivity to excited platinum levels of 192Pt and from 4.9% electron
capture to excited osmium levels of 192Os. At this
radioactivity of 192Ir, it emits a number of deexcitation lines of gamma
radiation in the range of 130-1380keV with significant
peaks 296keV (29%), 308 (30%), 316keV (83%), 468 (48%) and
604+613keV (8.2%). Several other very faint gamma lines of higher
energies are visible in the spectrum only at high magnification.
Conversely, at the beginning of the spectrum, the lines Ka,b of
the characteristic X-rays of osmium and platinum 61-78 keV are
visible, arising from deexcitation transitions in the envelope of
these daughter atoms after electron capture of the electron from
the K shell and after internal conversion of gamma
radiation.
Iridium-192 is used as an intense source
of gamma radiation with medium
energy in defectoscopy (§3.3,
part "Radiation
defectoscopy") and in brachytherapy
(§3.6, part "Brachyradiotherapy"). These high activity
emitters (GBq-TBq) are prepared by a nuclear reaction of 191Ir (n, g) 192Ir by irradiating
metallic iridium (wires, plates, pellets) with neutrons
in a nuclear reactor.
Conversion scheme and gamma spectrum of
iridium 192Ir
Thalium
Thalium Tl 81 is a soft white shiny metal from group III.B, which
occurs in nature only in compounds and relatively rarely
(approximately 0.05-2 mg/kg in the earth's crust). It has two
stable isotopes, 203Tl (29.5%) and 205Tl (70.5%). Of the radioactive isotopes of thallium, it
is worth mentioning the natural 208Tl (as a decay product of thorium-232) and especially
the artificially produced 201Tl :
Thalium
201 Tl
is an artificially produced radionuclide, which with a half-life
of 3 days is converted by electron capture EC to
201Hg.
From 21% it is to the baseline state, in 41% to the excited level
of 167keV and in 13% to the level of 32keV. During deexcitation,
gamma photons with energies of 135keV (2.6%) and 167keV (10%) are
emitted. However, the main component of the photon radiation 201Tl is the characteristic
X-rays of mercury, arising during the transitions of
electrons in the envelope after K-capture: Ka »
68-71 keV (73.7%) and Kb » 79-83 keV (20.3%), soft X
radiation of 8-15keV (43%) is of less importance. As with any EC
radionuclide, in the radiation of 201Tl are significantly present Auger electrons,
mainly with energies of 53-58keV (100%), 64-68keV (55%), 75-83keV
(7.6%), also low-energy electrons 5-15keV (> 50%).
The isotope thallium-201 is prepared by irradiating natural thallium
(or enriched in the isotope 203Tl) with protons in a cyclotron by a nuclear reaction of 203Tl (p, 3n) 201Pb, followed by
radioactive conversion of 201Pb with a half-life of 9.4 hours by electron capture on
the resulting 201Tl (an alternative option for the
preparation of intermediate 201Pb is the reaction 205Tl (p, 5n) 201Pb).
Note: It is
difficult to obtain absolutely pure 201Tl by these reactions - depending on the energy of the
irradiating protons, one or more neutrons are often ejected. In
most cases, therefore, the preparation also contains radionuclide
impurities of neighboring thallium isotopes 200,202Tl - see the
lower part of the figure of the 201-Tl spectrum.
Radionuclide 201Tl in the form of
chloride is used in nuclear medicine for scintigraphy of
myocardial perfusion *), as an analogue of potassium, in
a one-day protocol - §4.9.4 "SPECT myocardial perfusion". 201T1 cations accumulate in cardiac muscle cells similarly
to potassium ions. After application of radio indicator 201T1, areas of cardiac
muscle with reduced blood flow are displayed on the scintigram as
hypoactive areas.
*) 201Tl
was widely used especially in the 80s-90s. years. Its
disadvantage is the low energy of the dominant X-rays, which
causes poorer resolution and significant absorption in the
tissue; also a considerable radiation load (to which significantly represented Auger electrons also
contribute). Thalium has now been extruded
by 99mTc-
labeled isonitriles, with which much higher quality images are
obtained with a significantly lower radiation exposure. In the
near future, however, one may expect a partial
"renaissance" of thallium in connection with the
introduction of semiconductor CZT cameras (§4.2., part "Alternative physical principles of
scintillation cameras", passage
"Semiconductor cameras"), which have a higher
sensitivity for photon X radiation » 73keV of 201Tl.
Conversion scheme and gamma spectrum of thallium 201Tl.
Above: Pure
thallium-201. |
Bottom:
201-thallium prepared in a cyclotron by a nuclear
reaction
[203Tl(p,3n)201Pb; 201Pb(EC,T1/2=9,3hour)®201Tl]
often contains about 0.1% of radionuclide
impurities 200,202Tl .
In the gamma spectrum of such a preparation, in addition
to the basic peaks of 201Tl, weaker peaks with higher energies are
visible after magnification:
367,579,828,1205 keV from the 200Tl contaminant and peaks of 439, 520 keV from 202Tl. |
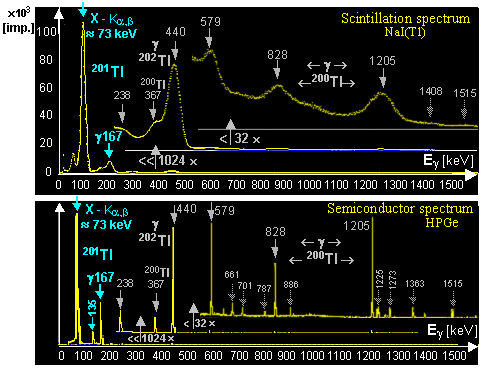 |
Lead,
Bismuth
The heavy metals lead and bismuth are the last elements of
Mendeleev's periodic table that still have stable
isotopes (bismuth is only a
"practically" stable isotope, see below). They are also end members of the radioactive decay
series of thorium, uranium and transuranes (see
"Natural radionuclides" above).
Lead Pb
82 (Plumbum)
is a soft low-melting metal (melting point 327.5 °C), which
is relatively rare in nature (approx. 0.05-2 mg/kg in the earth
's crust) *). It is mostly found in compounds such as lead
sulphide (galenit), lead sulphate and lead carbonate. It is
classically used in the production of batteries, lead glass,
ammunition. In the field of radiation physics, its high density
and absorption capacity for X and gamma radiation is important -
it is used as an effective shielding material (§1.6, section "Absorption of radiation in substances", passage "Shielding of ionizing
radiation").
*) However, the lead content is still
slightly higher than, according to cosmic
nucleogenesis, would correspond to its high proton number. This
is because lead isotopes are the end product of the radioactive
decay series of uranium and thorium (see Fig.1.4.1 ), so that over the course of billions of years, the
lead content in the earth's crust gradually increased.
Lead has 4 stable
isotopes: 204Pb (9.4%), 206Pb (24.1%), 207Pb (22.1%), 208Pb (15.4%). From the radioactive isotopes of lead worth
mentioning mainly three :
Lead 210 Pb
with a half-life of 22.3 years converts beta- -radioactivity to 210Bi - 20% in the
basic state, in 80% of the excited state 46.5keV. Max. beta
energy is relatively low, 63keV. In nature, it is constantly
emerging as one of the products of gradual transformation 238U in the uranium
decay series (Fig ....). In closed uranium-containing materials,
such as rocks, the 210Pb isotope is in permanent radioactive equilibrium with 238U and its daughter
isotopes.
Lead 214 Pb
with a half-life of 26.9min. converts by beta- -radioactivity to 214Bi, mainly to
excited levels of 295 and 362 keV, during the deexcitation of
which gamma radiation with energies of 352, 195 and 242 keV is
emitted - see below the spectrum of Radium 226 Ra. Like
214Bi in
nature is constantly formed as one of the products in the uranium
decay series (Fig ....). In closed materials containing uranium,
such as rocks, the isotope 214Pb is in permanent radioactive equilibrium with 238U and its daughter
isotopes.
Bismuth Bi
83 (Bismuthum), also called vismuth, is a shiny silvery
pinkish crystalline low-melting metal (melting
point 271 °C), rarely present in nature
(approx. 0.2 mg/kg in the earth's crust), as corresponds to its
high proton number. It is used for the preparation of low-melting
alloys such as Wood's metal (50%
Bi, 25% Bb, 12.5% Sn, 12.5% Cd), which has
a melting point of 60.5 °C. Due to its high density and
absorption capacity for X-rays, Bi is sometimes used in X-ray
diagnostics in shielding veils for radiation protection.
Natural bismuth is formed by the isotope 209Bi,
which was until recently considered stable. In
2003, however, it was found that this isotope is weakly
radioactive - with an extremely long half-life of 1.9.1019 years, it is subject
to alpha-radioactivity. From a practical point of view, however,
natural bismuth appears to be non-radioactive (300 kg of bismuth would have an activity of 1Bq), with radiation deep beneath the natural background. Of
the (really) radioactive isotopes of bismuth, three natural and
two artificial radionuclides are partly important :
Bismuth 212 Bi
occurs in nature as one of the products of the thorium decay
series (....). With a half-life of 60.5min. from 36% converted by
a-radioactivity
to 208Tl
and 64% by b-
-radioactivity to 212Po.
Bismuth 213 Bi
is converted from 97.8% by b --radioactivity
to 213Po
and from 2.2% by a-radioactivity at 209Tl .
Isotopes 214,213Bi are experimentally used in nuclear medicine for
radioimmunotherapy (§3.6, part "Radioisotope
therapy" passage "Radioimmunotherapy").
Bismuth 214 Bi
is one of the ephemeral but important products of decay series of
uranium (...). With the relatively short half-life 19.8 minutes
was converted by a complex decay scheme mainly beta- -radioactivity in a number of excited
level and the ground state of polonium
214Po, slight extent (0.021%) also
alpha radioactivity to excited levels
210Tl. When deexcitation (especially levels 214Po) a number of gamma radiation
energies are emitted, of which 609.3 keV (45.5%), 665 (1.5%), 768
(4.5%), 1120 ..... have a significant share -
see figure Ra -226 left.
Significant gamma-lines 214Bi can be observed
in uranium and radium (if it is in
equilibrium with its daughter nuclides - see below Radium 226 Ra) and also in most natural samples, where this
radionuclide enters with radon gas (see,
for example, the above figure 1.4.2).
Thorium,
Uranium, Radium, Actinium, Radon
Of tje heavy nuclei uranium groups, which are
all a -radioactive,
are especially important following nature
(primary) radionuclides of thorium and uranium.
Thorium Th
90 is a silvery white metal that is quite abundant in the
earth's crust (9.6 mg/kg) in mineral compounds. It has no stable
isotope, in terrestrial nature it occurs mainly weakly
radioactive thorium-232 :
Thorium 232 Th
Thorium 232Th
90 is one
of the most widespread natural radionuclides
contained in the rocks of the earth's crust (along
with 40K), its content is about 9 mg/kg. With a very long
half-life T1/2 = 14,02.109 years, it decays by alpha-radioactivity
to radium 228Ra and then by the whole decay series according to the
left part of Fig.1.4.1.
It does not yet have direct radiation use, but in the future it
may be a potential fissile material in thorium-uranium
cycle in propagation nuclear
reactors - §1.3, part "Fast FBR
propagation reactors". Due to the low specific activity and the impossibility
of use for direct chain fission, thorium-232 can be considered as
an almost non-radioactive material from the point of view of
nuclear physics and radiation protection, with neglible (or zero) risk.
Thorium can be used as a material for effective shielding
of ionizing radiation. Furthermore, 232Th is sometimes used in technical practice as an
additive to lamp electrodes, where ionization of
the gas charge by alpha particles facilitates the ignition of an
electric discharge at a lower voltage (§3.7,
passage "Radioactivity in lamps"). Another use of thorium
(230-Th) is in uranium-thorium radiometric dating (see "Radiometric dating"
above).
Thorium
227 Th
is an artificially produced radioisotope for use in biologically
targeted radionuclide therapy. With a half-life of T1/2 =
18.7 days, by alpha-radioactivity is
converted to radium 223Ra - in 25% to baseline, 24% to
61.5keV, 20% to 286keV, 5% to 329keV, in 8% to 334kev, with a
lower representation on a number of other excited states. During
deexcitation, a number of gamma photons are emitted, mainly
energies of 50keV (8%), 236keV (12.4%) and 256keV (7%), weaker
are 300keV (2.3%) and 330keV (2.7%). The energies of
alpha-particles range from 5.7-6 MeV, depending on the excited
levels. The daughter 223Ra is then again a-radioactivive, with a half-life of 11.4 days converts
to radon-219 and further through the whole decay chain
of short-term isotopes to stable lead-207 (cf.
the picture below in the passage "Radium
223 Ra"), with the emission of many
other energies of alpha particles, beta electrons and gamma
photons :
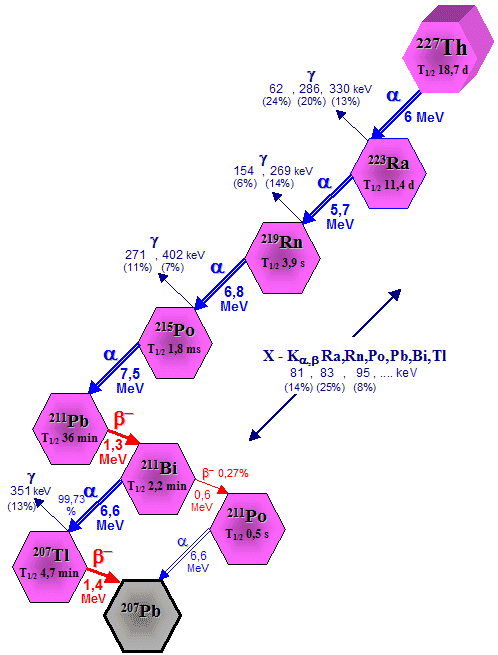 |
As
is typical for heavy uranium nuclides (and
higher), even 227 Th is converted by a whole decay
series : 227Th(18,7d.; a) ® 223Ra(11,4d.; a) ® 219Rn(4s.; a) ® 215Po(1,8ms.; a) ® 211Pb(36,1min.; b-)
® 211Bi(2,2min.; a) ® 207Tl(4,8min.; b-)
® 207Pb(stable).
During the first conversion of 227Th
(to 223Ra radium), alpha
radiation with energies of 5.7-6 MeV and gamma
radiation with energies of mainly 50, 236 and 256
keV are emitted.
When the radioactivity of daughter nuclides decay
chain is emitted, then a number of other energy
alpha particles, beta and gamma photons, in
particular: from 223Ra it's a 5.6
5,7MeV and gamma rays with energies of 154 and
especially 269 keV; of 219Rn is it a 6.4
and 6.8 MeV and g 271 and 402
keV; of 215Po it a 7,4MeV;
of 211 Pb it is beta 540 and
1372 keV and weak g 404 and 832
keV; from 211Bi it is a 6.3
and 6.6 MeV and g 351keV; of 207Tl , beta max. energy
1423keV and weak gamma 898keV are emitted.
Secondary branch 211Bi (b-) ®
211Po due to its low proportion (0.27%), it
has no practical significance.
The 227-Th decay chain functions as an
"in vivo radionuclide generator"
- see "In vivo
generators"
above.
A simplified conversion scheme of 227Th
to 223Ra is drawn below in the left part of the
figure.
The measured spectrum of gamma
radiation emitted by all nuclides from the entire
227-Th decay series is shown below in the right
part of the figure. e |
|
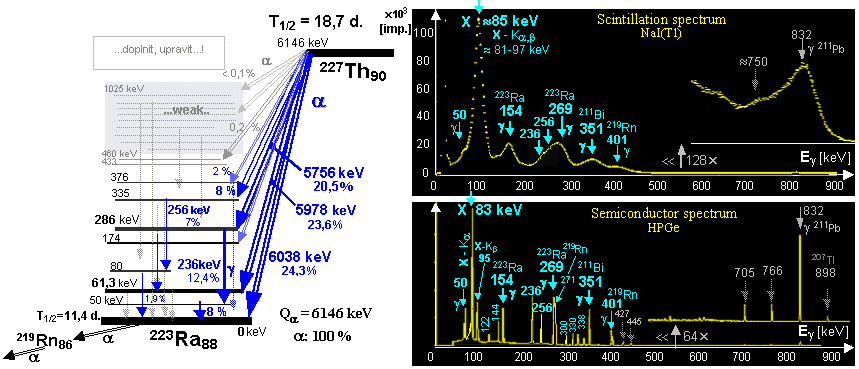 |
Conversion
scheme of 227Th thorium (first
conversion to radium-223) and the
total gamma spectrum of 227Th and its decay products.
The spectrum was measured with a
hermetically sealed preparation, from which 219Rn radon gas
could not escape. The spectrum therefore contains gamma
lines and all daughter radionuclides of the decay series,
with which 227Th is in radioactive equilibrium. The gamma
lines derived from the conversion of 227-Th to 213-Ra are
given in the spectrum without labeling (only with energy
value), for photopeaks from secondary transformations in
the decay series, the corresponding radionuclides from
which they originate are listed (especially 223Ra, 219Rn, 211Bi, 211Pb and 207Tl). |
One complete radioactive
transformation of the 227Th nucleus in the entire decay series
to a stable 207Pb releases a total nuclear energy Q = 36.132
MeV, which is carried away by about 95% by five
alpha particles, by 3% by beta + neutrons and by about
1% gamma photons *) and X-rays.
*) This seemingly small percentage of
photon radiation is due to the energy recalculation with respect
to high-energy a-particles. In fact, the absolute number of gamma and X
photons emitted is relatively high, for energies
in the range of 60-400keV represents about 96%! The most
represented X and gamma-energies are: X 81 (14%), 83 (25%) and
95keV (8%), gamma 154 (5.8%), 269 (14%), 351 (13%) and 402keV
(7%).
For thorium 227Th and daughter products of the decay series, mainly
emitted alpha radiation with energies of 5.4-7.4
MeV (a total of 5 alpha
particles /1 conversion of 227Th) is used in nuclear medicine for
biologically targeted radionuclide therapy.
Gamma
radiation of 227Th and its daughter
nuclides are practically does not manifest in therapy, but can be
used for gammagraphic monitoring of the
distribution of radiopharmaceuticals in the body. Emission of gamma
radiation during therapy allows simultaneous scintigraphic
imaging of tissues and organs into which the
therapeutic preparation has penetrated. On these scintigraphic
images we can assess (visually, or even
quantitatively) the degree of desirable
uptake of radiopharmaceuticals in tumor foci and unwanted
accumulation in healthy tissues and critical organs - to monitor
the course of radioisotope therapy.
Chemical properties of thorium (oxidation
state +4) allow, with the
help of a macrocyclic ligand of the DOTA type, chelating binding
of 227Th
to monoclonal antibodies to form radioimmunoconjugates
that are specifically taken up in tumor cells (the structure and properties of monoclonal antibodies
are discussed in more detail in §3.6, section "Targeted biological therapy - monoclonal antibodies"). High energy a particles capable
emitted during radioactive transformations, they can effectively destroy
the tumor cells. In the laboratory trial phase, targeted
immunotherapy using alpha- labeled thorium
monoclonal antibodies is in lymphomas 227Th-rituximab or anti-CD22, in acute myeloid leukemia 227Th-conjugate CD33
lintuzumab.
A certain problem may be the release
of daughter radium-223 from the chemical bond in the
radioimmunoconjugate due to the backscattering of the
nucleus during alpha-particle emission (§1.2, passage "Backscattering of the nuclei") and differences in the
chemical properties of the radium (oxidation number 2). If
daughter radium escapes from the target tissue during its
transformation (T1/2 = 11.4 days), the effectiveness of
the therapy is reduced by 4 energetic
alpha-particles from the radium-223 decay series. The released 223Ra can then be taken
up in other tissues (eg bones) and cause unwanted radiation exposure there.
Preparation: Isotope 227Th is artificially
prepared by irradiating radium-226 with neutrons to form
radium-227: 226Ra (n, g) 227Ra, followed by conversion by beta-- radioactivity 227Ra (b-, T1/2 =
41min.) ® 227Ac to actinium-227,
which with a half-life of 21.8 years is converted to the
resulting thorium-227: 227Ac (b-, T1/2 = 21.8y.) ® 227Th. Due to the long half-life of actinium-227, 227Th can be
continuously obtained by elution from a 227Ac/227Th generator.
Uranium U
92 is a very heavy (19 g/cm3) metallic element of silver-white color from the group of
actiniodes. It acquires a dark gray color in air due to
oxidation. It was isolated in 1841 and named after the then newly
discovered planet Uranus. It was used to color glass. In
1896, H.Becquerel discovered previously unknown penetrating
radiation in uranium-containing minerals - he discovered
radioactivity (§1.2 "Radioactivity"). Uranium has no stable
isotope, all 28 known isotopes are radioactive.
The natural metallic element uranium U92 consists mainly of the isotopes 235U (0.7204%) and 238U (99.2742%), which are important natural
primordial radionuclides; there is also a trace amount of 234U (0.0054%), which
is formed as a secondary intermediate of the decay series 238U.
Uranium 235 U
decays with alpha-radioactivity to thorium 231Th with a long
half-life T1/2 = 7.04.108 years and then the
whole decay series according to the middle part of Fig.1.4.1. By absorbing the neutron, the 235U nucleus splits into two lighter
nuclei. Uranium-235 is the basic fissile material
in nuclear reactors, the fission of which
generates a large amount of energy in nuclear power plants;
nuclear reactors also serve as a powerful source of
neutrons for many applications of nuclear physics and
chemistry. For details, see §1.3 "Nuclear
reactions", section "Fission of atomic nuclei".
Uranium 238
U
with a very long half-life T1/2 = 4.47.109 years decays by alpha-radioactivity to
thorium 234Th
and then by the whole decay series according to the right part of
Fig.1.4.1. Due to its longer
half, it is 238U the most common type of uranium
in nature. It can also be used as fissile material, but not
directly, but via plutonium, which is formed in the
nuclear reactor from 238U by neutron absorption (§1.3,
section "Fast FBR
propagation reactors"). Uranium-238 (as so-called "
depleted uranium ") has only
relatively low radioactivity (see the table
in the section "Relationship between half-life
and activity " §1.2 "Radioactivity") and is sometimes used as an effective shielding
material for high-energy gamma radiation.
Uranium's own
alpha-radioactivity produces relatively little gamma radiation.
In g-specrum,
we can see from uranium only not very significant lines 185keV
and 205keV from the conversion of 235U to metastable levels 231Th. Most gamma-radiation of uranium comes from daughter
radionuclides of the decay series, mainly from thorium 234Th (63keV, 93keV),
then from radium-226 (185keV) and mainly from lead 214Pb and bismuth 214Bi (214, 295, 352,
609 keV and many others...).
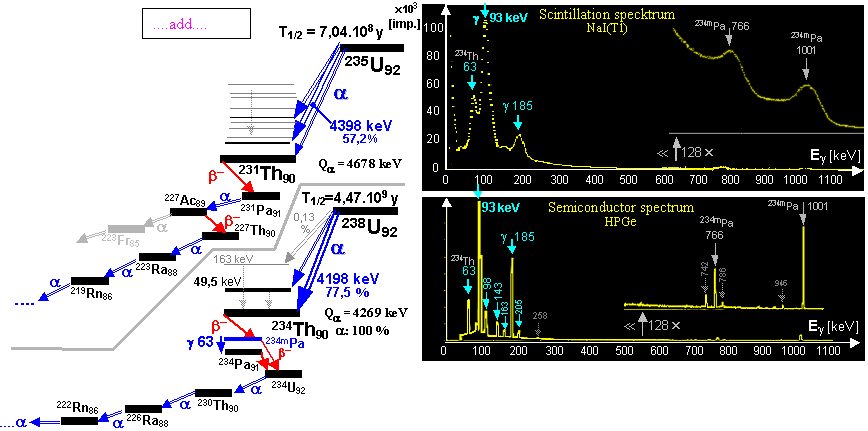
Simplified conversion scheme of uranium 235U and 238U and gamma spectrum of natural uranium and some of its
decay products.
The gamma spectrum was measured from a
sample of loose 0.3 mm thick uranium metal sheet. A series of
daughter radionuclides in the decay series was interrupted here
for radon 219.222Rn, which, due to its gaseous state, escapes smoothly
from the sample. Therefore, there are no gamma lines in the
spectrum of other radionuclides of the decay series, eg 214Pb and 214Bi, which can be
seen, for example, in the spectrum of a sample of encapsulated
radium (see "Ra-226.gif" below), or in
samples of the natural environment (e.g. "NaturalRadioactivity.gif").
Uranium 234 U
decays with a half-life T1/2 = 245.5 thousand years by alpha-radioactivity
to thorium 230Th and then by a 238-uranium decay series to lead-206.
It is formed in small quantities as part of the 238U decay
series. It has no practical significance, it is not a fissile
material.
The last somewhat technically significant isotope of
uranium is man-made :
Uranium 233 U ,
which decays with alpha-radioactivity
to thorium 229Th with a half-life of T1/2 = 159.2 thousand years (and then a
neptunium decay series to Bi-209 - shown below in the
passage "Actinium 225 Ac", in
the picture on the top left). Uranium-233
is formed from thorium 232Th by neutron absorption in the reaction 232Th90(n,g)233Th90®(b-;12min)®233Pa91®(b-;27
days)®233U92. Uranium 233U can potentially serve as fissile material in thorium
fuel cycle reactors (Fast Propagation Reactors FBR).
The decay products of thorium and uranium are a number
of other radioisotopes (they form the
so-called decay series described above, Fig.1.4.1 ), the most important of which is radium :
Radium Ra
88
is an important radioactive element associated with the history
of radioactivity in the early 20th century. Radium is a
silvery-white, heavy and highly reactive alkaline earth metal.
All its isotopes are radioactive. In nature, four
isotopes of radium occur in trace amounts: 223Ra (T1/2 =
11.4 d.), 224Ra (T1/2 =
3.64 d.), 226Ra (T1/2=
1602 years), 228Ra (T1/2 =
5.75 years). They come from the decay series of uranium and
thorium. Radium isotopes with alpha-radioactivity are converted
to the corresponding radon gas isotopes and then through
a whole 5-6 member decay series to a stable lead isotope (with the exception of 228Ra in the thorium series, which is converted by a
10-membered series comprising radium-224 and radon-220).
Radium
226 Ra
The most important isotope of radium is 226Ra, discovered and isolated from
uranium ore in 1898 by M. and P. Curie. 1 gram of 226Ra, isolated by the
heroic efforts of Mrs. M.Curie and their collaborators, served
for a long time as a standard of the former unit
of activity 1 Curie (1Ci). With a half-life of T1/2 =
1602 years, 226-radium is converted by alpha-radioactivity
to radon 222Rn (and then by a whole decay
series to a stable lead of 206Pb). In 94.4% takes place a conversion to the
basic state of radon, 5.5% in the excited state
186keV, three other higher excited levels are
underrepresented.
226Ra itself is a mixed
a + g emitter (Ea =
4.78MeV, Eg = 186keV), but in reality it is
in equilibrium with its daughter isotopes in the decay series. In
the spectrum of gamma radiation 226Ra we see our own peak 186keV, but the lines of daughter
products 214Pb and 214Bi are much more pronounced (with higher energies: 295, 352, 609, 768, 1120, 1238,
1378, 1764, 2204, 2448 keV). Radiophores 226Ra were previously
used in brachyradiotherapy (§3.6,
part "Brachyradiotherapy"), are now extruded mainly
by iridium-192 (see"Iridium" above).

Conversion scheme of 226Ra radium and gamma spectrum of 226Ra and its decay products. In the section in the left
part of the figure there are simplified decay schemes of
important radionuclides 214Pb and 214Bi from the decay series radium-226.
The spectrum was measured with an
encapsulated 226Ra standard from which 222Rn radon gas could not escape, so that 226Ra is in radioactive
equilibrium with all its daughter products. Therefore, in the
spectrum (in contrast to the above-mentioned uranium spectrum),
the gamma lines of all daughter radionuclides of the decay
series, especially 214Pb and 214Bi, are displayed.
Radium
223 Ra
is (unlike previous natural nuclides) artificially produced radioisotope for
radionuclide therapy in nuclear medicine (naturally isotope 223Ra present in very small quantities as one of the decay
products of uranium 235U in the uranium-actinium decay chain - Fig.1.4.1). 223-radium is converted by alpha-radioactivity
*) with a half-life of 11.43 days to radon 219Rn (and then by a decay series to stable lead 207Pb, see below). Only 1% is converted to the ground state, in most
cases it is a series of excited states 219Rn. The most represented are conversions to levels with
energies of 127keV (9.5%), 154keV (52.5%), 269keV (24.2%), 338keV
(9.2%), during the deexcitation of which gamma radiation is
emitted (the largest proportion here they
have energies of 154 and 269keV, the weaker is 324keV). Alpha radiation from 223Ra conversion has energies in the range of 5.5-5.7MeV.
Additional energies alpha, beta and gamma are emitted from the
daughter products of the decay series, see below - cf. also the picture above in the passage "Thorium
227 Th".
*) A little interest :
223Ra was the first isotope for which a new radioactive
transformation was discovered in 1984 with the emission of
particles heavier than alpha-particles - "carbon
radioactivity" with the emission of nuclear 14C: 223Ra ® 209Pb + 14C (see §1.2, part "Exotic species of radioactivity",
passage "Radioactivity higher than a -helium").
However, this process is very weak, at the limits of
measurability, the ratio of the number of emissions of 14C and alpha emissions
(ie 4He)
is 6.4.10-10.
Preparation: Isotope 223Ra is artificially
prepared by irradiating radium-226 with neutrons
to form radium-227: 226Ra (n, g) 227Ra, followed by conversion by beta --radioactivity 227Ra (b-, T1/2 =
41min.) ® 227Ac to actinium-227,
which with a half-life of 21.8 years is converted via thorium-227
to the resulting radium-223: 227Ac (b-, T1/2 = 21,8y.) ® 227 Th (a, T1/2 =
18,7d.) ® 223Ra. Due to the long
half-life of actinium-227, 223Ra can be continuously obtained by elution from a 227Ac/223Ra
generator (selective elution is
performed with a solution of about 0.1 mol. HCl or HNO3).
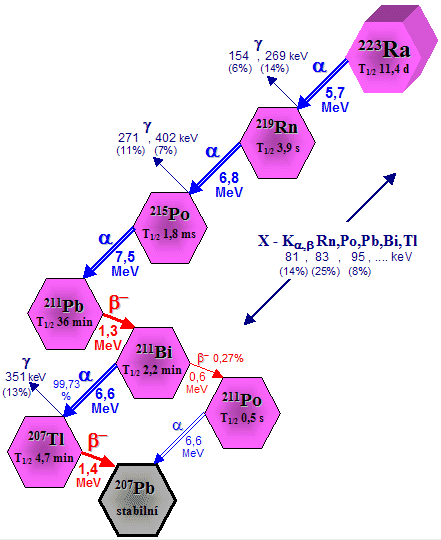 |
As
is typical for heavy uranium nuclides (and
higher), even 223Ra is transformed by the whole decay
series :
223Ra(11,4d.; a) ® 219Rn(4s.; a) ® 215Po(1,8ms.; a) ® 211Pb(36,1min.; b-)
® 211Bi(2,2min.; a) ® 207Tl(4,8min.; b-)
® 207Pb(stab.)..
During the first conversion of 223Ra
(to radon 219Rn), alpha
radiation with energies of 5.6 and 5.7 MeV and
gamma radiation with energies of especially 154
and 269 keV are emitted.
During the radioactivity of the daughter
nuclides of the decay series, a number of other
energies of alpha, beta and gamma photons are
emitted, in particular: from
219Rn
it is a 6.4 and 6.8 MeV and g
271 and 402 keV; of
215Po
it is a
7,4MeV; of 211Pb it is beta 540 and 1372 keV and weak g 404
and 832 keV; from 211Bi it is a 6.3
and 6.6 MeV and g 351keV; of 207Tl , beta max. energy
1423keV and weak gamma 898keV are emitted.
Secondary branch 211Bi (b-) ®
211Po due to its low proportion (0.27%) has
no practical significance.
The 223-Ra decay chain functions as an
"in vivo radionuclide generator
" - see "In vivo
generators"
above.
The measured spectrum of gamma radiation
emitted by all nuclides from the entire 223-Ra
decay series is shown below in the right part of
the figure. e |
|
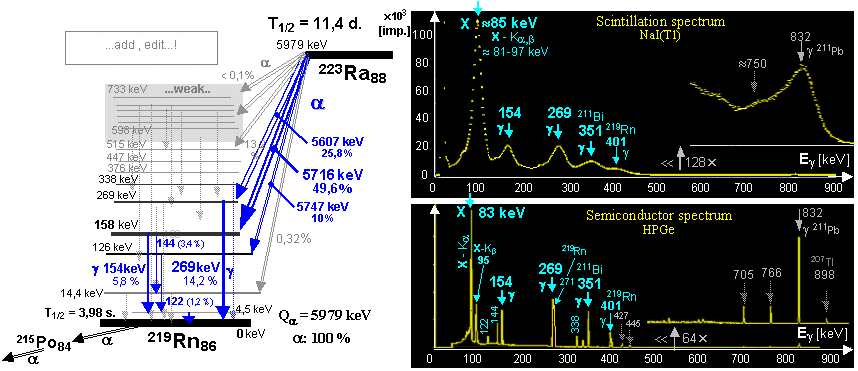 |
Conversion
scheme of 223Ra radium (first
conversion to radon-219) and the
total gamma spectrum of 223Ra and its decay products.
The spectrum was measured with a
hermetically sealed preparation, from which 219Rn radon gas
could not escape. The spectrum therefore contains gamma
lines and all daughter radionuclides of the decay series,
with which 223Ra is in radioactive equilibrium. The gamma
lines derived from the conversion of 223-Ra to 219-Rn are
given in the spectrum without labeling (only with energy
value), for photopeaks from secondary transformations in
the decay series, list the corresponding radionuclides
from which they originate (especially 219Rn, 211Bi, 211Pb and 207Tl). |
One complete
radioactive transformation of the 223Ra nucleus in the whole decay series to
a stable 207Pb releases a total nuclear energy Q = 29.986
MeV, which is carried away by about 95% by four
alpha particles, by 3% by beta + neutrons and by about
1% *) photons gamma and X-rays.
*) This seemingly small percentage of photon radiation is due to
the energy recalculation with respect to high-energy a-particles. In
fact, the absolute number of gamma and X photons emitted is relatively
high, for energies in the range of 80-400keV represents
about 96%! The most represented X and gamma-energies are: X 81
(14%), 83 (25%) and 95keV (8%), gamma 154 (5.8%), 269 (14%), 351
(13%) and 401keV (7%).
From the radium 223Ra and daughter products of the decay series use mainly
emitted alpha-radiation with energies of 5.4-7.4
MeV (a total of 4
alpha-particles/1 conversion of 223Ra) in nuclear
medicine for radioisotherapy of bone metastases,
which often occur in breast and prostate cancer (§3.6, part "Radioisotope therapy", passage "Radionuclide therapy
of tumors and metastases"). Beta electrons are of negligible importance. Radium
has a calcium-like chemical and biological behavior, so that 223Ra accumulates in
metabolically active bone tissue in hydroxyapatite crystals. It
is therefore taken up in bone metastases with increased
osteoblastic activity, where it unfortunately shows only
a palliative effect in the osteoblastic margin around
the metastasis - alpha-particles practically do not penetrate
into the tumor tissue itself (see below
"Critical
note : 223Ra is not a suitable therapeutical
radionuclide!").
The manner in which
the parent 223 Ra is converted to a number of shorter radionuclides
after its accumulation in the target tissue is a typical example
of the aforementioned in vivo radionuclide generator .
Gamma radiation 223Ra and its daughter
nuclides are practically not used in therapy, but can be used for
gammagraphic monitoring of the distribution of
radiopharmaceuticals in the body. The positive significance of
the relatively large proportion of gamma radiation also lies in
the possibility of easily measuring the activity
of 223Ra preparations in conventional activity meters with an
ionization chamber (§2.3 "Ionization chambers"), using the appropriate
gamma constant calibration.
Critical
note : 223Ra is not a
suitable therapeutical radionuclide !
The basic condition for the usability of a given radionuclide in
nuclear medicine is the possibility of chemical
attachment of its atom to a suitable biochemically
targeted molecule, which ensures "transport" of the
radionuclide to target tissues and organs for diagnostic imaging
or therapeutic effect. This chemical attachment can be direct
(for simple molecules) or via chelating agents
(especially for monoclonal antibodies). A sufficiently strong
bond, direct or with a chelating agent, depends mainly on the oxidation
number (valence) of the atoms of the given element.
The element radium, and thus 223Ra, has an oxidation number of 2 and suitable chelating
agents are not available for it. So radium is not
promising for the formation of 223Ra-radioimmunoconjugates! In contrast,
the radionuclide atoms thorium 227Th or actinium 225Ac, with oxidation number 4, are able
to form highly stable complexes with chelators and are therefore
suitable for biologically targeted radionuclide
therapy.
Despite these facts, the therapeutic preparation 223RaCl2 Alpharadin
was developed with radio 223Ra (developed by Nanovector with the utmost care ...),
later renamed by Bayer Xofigo, for palliative therapy of
bone metastases. Corporate advertising also spoke of a curative
effect, which was not confirmed. And in the end, the palliative
effect turned out to be practically the same as in the
preparations used for many years with radionuclides strontium 89Sr (Metastron)
or samarium 153Sm (with phosphate ligand EDTMP), which are several times cheaper and tested. The use of
223Ra for
the treatment of bone metastases in prostate cancer is now
meaningless, given that there are potent 177Lu or 225Ac-labeled anti-PSMA monoclonal antibodies that are able
to have a curative effect not only on bone
metastases but also on tumor foci in all other tissues and
organs, even at an advanced stage.
The enforcement of the 223Ra radium was wrong from the very
beginning - this is how we reflected it at our workplace. When
most colleagues in the field of nuclear medicine realize this, 223Ra will probably be definitively
abandoned ...
Actinium Ac 89 is a soft silvery white metallic element, highly radioactive,
has no stable isotope. In nature, two isotopes of actinium occur
in trace amounts: 227Ac (T1/2 =
21.77 years) and 228Ac (T1/2 =
6.15 hours), as part of the decay series of uranium-235 and
thorium-232. They decay by alpha (or branched beta-alpha)
radioactivity to the corresponding francium (or thorium) isotopes
and further by a 4-6 member decay series to stable lead isotopes.
The name of the element originated from its strong radioactivity (Greek aktis = ray). The
isotopes of actinium 206-234Ac are known in nuclear physics. The most important of
these - for radionuclide therapy in nuclear medicine - is the
artificially produced isotope actinium-225 :
Actinium
225 Ac
With a half-life of 10 days, it is
converted by alpha-radioactivity to francium 221Fr - in 50% to the
ground state (emission Ea = 5.83MeV), in other cases to a number of excited states
of francium (several tens of gamma photons
are emitted during deexcitation, from low energies approx. 10keV,
weaker peaks are up to 800keV). Further
radioactive transformations of 221Fr then continue through the entire decay series
up to bismuth 209Bi - it is part of the neptunium series :
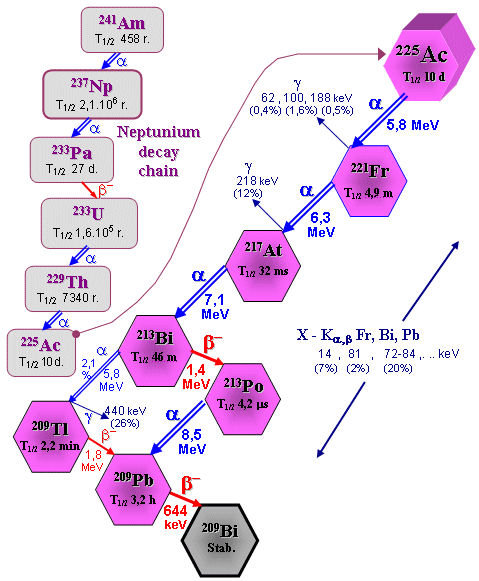 |
Actinium
225Ac is transformed by a whole decay
series of 6 main daughter nuclei :
(225Ac(10d.; a) ® 221Fr(4,8m.; a) ® 217At(32ms.; a) ® 213Bi(46m.; b-)
® 213Po(4ms.;
a) ® 209Pb(3,3h.; b-)
® 209Bi(stab.)) - picture on the left.
Note:
209Bi can be
considered stable here (a, T1/2 » 2.1019
years).
In
the first conversion of 225Ac
(to franicum 221Fr) alpha
radiation with an energy of 5.8 MeV and weak
gamma radiation of 63, 100, 110, 150 keV are
emitted, higher energies of 250-760keV have a
small representation.
During the radioactivity
of the daughter nuclides of the decay series, a
number of other energies of alpha, beta and gamma
photons are emitted: of 221Fr it is a
6.3 MeV and gamma radiation with energy mainly
218 keV; of
217At
it's a 7.06 MeV; from
213Bi
it is alpha 5.8MeV, beta 987keV and gamma 440
keV; of 213Po is alpha 8.4MeV; of 209Pb it is beta 644keV.
The
secondary branch 213Bi (a)
® 209Tl is not more important in applications
due to its low proportion (2.1%).
The 225-Ac decay chain functions as
"in vivo radionuclide generator
"- see above "In vivo
generators".
In contrast to thorium-227, 225Ac has the advantage that its decay
chain does not contain longer-lived daughter
radionuclides, so there is less risk of premature
leakage of radioactivity from the target tissue.
A simplified conversion scheme
of 225Ac to 221Fr and another decay chain is drawn
below in the left part of the figure.
The measured spectrum
of gamma radiation emitted by all nuclides from
the whole 225-Ac decay series is shown below the
right portion of fig.: ê |
|
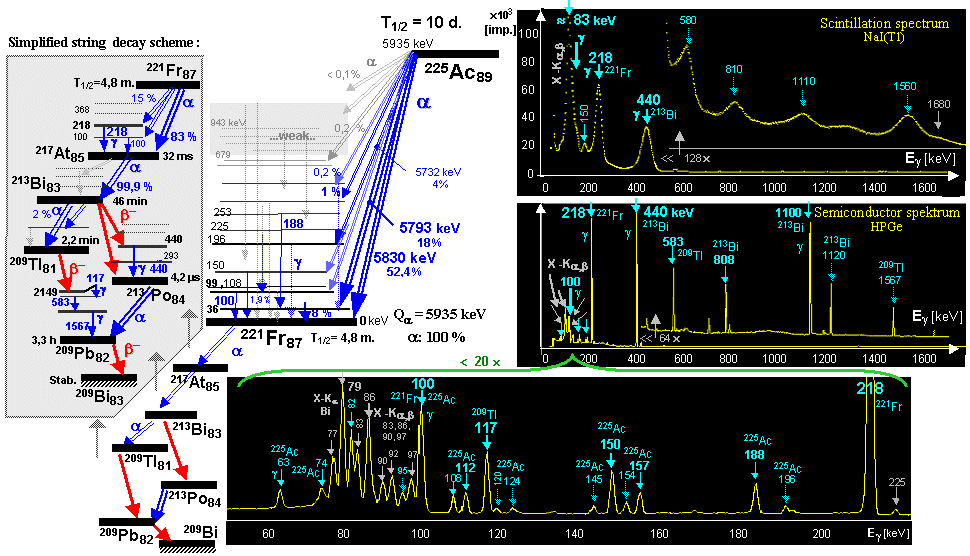 |
Conversion
scheme of actinium 225Ac (first conversion to
francium-221, simply next chain) and
the total gamma spectrum of 225Ac and its decay products.
The spectrum contains gamma lines
and all daughter radionuclides of the decay series, with
which 225Ac is in radioactive equilibrium. At the bottom
of the figure is an enlarged section of the spectrum of
the low-energy region of about 50-230 keV, where there is
a large inflation of gamma and X peaks.
Actinium-225 alone, when alpha-converted to 221-Fr,
produces relatively little gamma radiation (only faint
peaks of 63, 100, 150, 188 keV are visible in the
spectrum). Most gamma radiation of actinium preparations
comes from daughter radionuclides of the decay series,
mainly from 221Fr (218keV) and 213Bi (440keV). |
One complete
radioactive transformation of the 225Ac nucleus in the entire decay series
up to a stable 209Bi releases a total nuclear energy Q = 31
MeV, which is carried away by about 95% by four
alpha-particles, by 3% beta + neutrinos and by about 1%
gamma photons *) and X-rays.
*) This seemingly small percentage value of
photon radiation is due to the energy recalculation with respect
to high-energy a-particles. In fact, the absolute number of gamma and X
photons emitted is relatively high, for energies
in the range of 60-440 keV represents about 96%! The most
represented X and gamma-energies are: X 14 (7%), 81 (2%), 72-84
(20%) ...., gamma 62 154 (5.8%), 269 (14%), 351 (13%) and 401keV
(26%).
For actinium 225Ac and daughter products of the decay series, emitted alpha
radiation with energies of 5.8-8.4 MeV (a total of 4 alpha particles/1 conversion of 225Ac) is used in nuclear medicine for biologically targeted
radionuclide therapy. Radiopharmaceuticals labeled with 225Ac (including monoclonal antibodies, eg 225Ac-Trastuzumab or 225Ac-PSMA-617) are being tested for the treatment of leukemia,
lymphomas, neuroendocrine tumors, gliomas, melanomas, and are very
promising in the prostate ca - 225Ac labeling of antiPSMA.
Radiation of gamma
daughter nuclides 225Ac is practically not contribute in therapy, but can be
used for gammagraphic monitoring of the
distribution of radiopharmaceuticals in the body. Emission of gamma
radiation, especially energy 218keV, allows during
therapy, simultaneous scintigraphic imaging
tissues and organs into which the therapeutic preparation has
penetrated. On these scintigraphic images we can assess (visually, or even quantitatively)
the degree of desirable uptake of radiopharmaceuticals in tumor
foci and unwanted accumulation in healthy tissues and critical
organs - to monitor the course of radioisotope
therapy.
Preparation: Isotope 225Ac in larger
quantities is still difficult to obtain. It can be obtained as a
product of radioactive alpha + beta conversion of thorium 229Th (a, T1/2 = 7340 years) ® 225Ra (b-, T1/2 = 14.8 days) ® 225Ac - thorium-actinium 229Th
/ 225Ac generator, from which a smaller
amount of actinium has so far been obtained for experimental use
in nuclear medicine. Thorium-229 is gained from a -decay of uranium 233U, whose supplies
are very limited. For rutine production, actinium-225 can be
prepared in a cyclotron by irradiating radium-226 with protons (optimal energy 17MeV) by reacting
226Ra (p,
2n) 225Ac.
Actinium 227 Ac
With a half-life of 21.77 years, it is transform
branching by beta-
-radioactivity to 227Th (98.6%) and alpha-radioactivity to 223Fr (1.4%), then by
decay series.... It is produced by irradiating radium-226
neutrons in a nuclear reactor: 226Ra (n, g) 227Ra (b-, T1/2 = 41min.) ® 227Ac. Actinium 227Ac is used as the parent isotope for the generator
preparation of the above-described thorium 227-Th and radium 223-Ra.
Among the decay
products of thorium and uranium, gaseous
radioisotopes of radon occur :
Radon Rn
86 is the heaviest element in the group of rare gases. It
is radioactive, it has no stable isotope. In nature, three
isotopes of radon occur in trace amounts: 222Rn (T1/2 =
3.82 d.), 220Rn (T1/2 =
54.5 s.) and 219Rn (T1/2 =
3.92 s.). They come from the decay series of uranium and thorium,
specifically they arise during the alpha-radioactivity of radium
isotopes. They decay into the appropriate isotopes of polonium
and further by a decay series of 4-6 members into stable isotopes
of lead. The original name of radon was radio emanation
- springing, emanating from radium. And radon 220Rn was formerly
called thoron (it is part of the thorium
decay series -Fig.1.4.1
left).
Radon 222 Rn
Alpha-decay of radium-226 produces gaseous radon 222Rn,
which is further converted with a half-life of 3.83 days by alpha-radioactivity
to polonium 218Po, in 99.92% to the ground state, in 0.08% to the
excited level 510keV. Further radioactive transformations then
take place throughout the decay series according
to the right part of Fig.1.4.1. The gamma spectrum of radon-222 is almost identical to
the above spectrum of 226 Ra (except for the 186keV line). Secondary natural
radon-222 with its decay products is important from the point of
view of radioecology - see chapter
5, §5.2 "Biological effects of ionizing
radiation", section
"Sources of ionizing radiation".
Radons 220Rn in thorium and 219 Rn in uranium-actinium decay series due to their short
half-life are sufficient to decay before diffusion escapes from
radioactive material, so there is no need to consider in
radioecology.
Transurans
Of the artificially produced heaviest nuclei of the transuranic
group (elements heavier than uranium - they
are discussed in detail in §1.3, part "Transurans"), three radionuclides have
a practical application :
Plutonium 239 Pu
Plutonium 239 Pu with a half-life of T1/2 = 24100 years by alpha -radioactivity
decays to uranium 235U (to excited levels of 51.7, 13.4
and 0.007 keV; however, gamma radiation is almost not emitted
here, because the respective transitions are almost completely
subject to internal conversion, so that conversion and Auger
electrons are emitted instead). Further
transformations are taking place throughout
uranium-actinium decay series (middle
part of Fig.1.4.1). Plutonium-239 is formed in a nuclear reactor by
neutron fusion from uranium 238U: 238U (n, g) 239U ® (b- ; 25min) ® 239Np ® (b-
; 2.3 days) ® 239Pu. It is an
effective fissile material (similar to uranium 235U). In addition to
misuse for nuclear weapons, it is used in special nuclear
reactors (§1.3, section "Fast breeding
reactors FBR").
Americium
241 Am
Americium 241 Am decays with a half-life T1/2 = 458
years by alpha-radioactivity to neptunium 237Np (and then by the whole neptunium decay series - shown above in the passage "Actinium
225
Ac", in the diagram on
the top left). Alpha-decay of 241Am occurs mainly (in
84.6%) to an excited level of 59.6 keV of
neptunium, which emits 59.6 keV gamma radiation
when deexcited to the ground state, 23.6 keV gamma is weakly
represented. As a result of the internal conversion, the characteristic
X-radiation L is further emitted La,b of neptunium
11-22 keV. A large number of other excited levels of 237Np (33, 75-1014 keV) are saturated
with negligible intensity and have no significance for 241Am radiation.
241-americium is thus a mixed a
+ g emitter
: Ea = 5485keV, Eg = 59.6keV. Due to the very long half-life of the
daughter 237Np, the radiation of other members of the decay series
is negligibly low compared to americium.
The 241Am isotope is formed in a plutonium in nuclear reactor
by neutron fusion in reactions 239Pu (n, g) 240Pu (n, g) 241Pu ® (b-) ® 241Am.
Americium-241 is often used as a standard
soft gamma radiation 59.6 keV, as source of a particles
e.g. in the ionization fire detectors, mixed with beryllium using
reaction (a, n) as a laboratory source of neutrons (§1.6, section "Neutron radiation and its interactions").
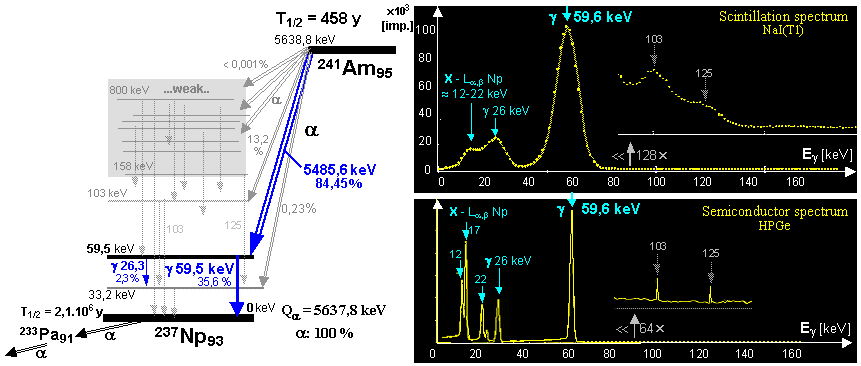
Decomposition scheme and gamma spectrum of americium 241Am .
Californium 252 Cf
is the heaviest radionuclide, which still has a practical
application. Californium 252Cf (T1/2 = 2.65 years) decays, in addition to a-radioactivity
(97%) to 248Cm, by spontaneous fission (3%) *), in
which neutrons are emitted (approx.
3.7 neutrons/1 fission) - it is therefore
used as an intensive neutron source, eg for
neutron activation analysis and for neutron capture
radiotherapy (chapter 3.6., section "Neutron Therapy"). Californium-252 is formed from a plutonium- 239Pu in nuclear
reactor by gradual multiple neutron capture, combined with beta-
transformations.
*) Note: From the
point of view of neutron production, perhaps even more
interesting would be the isotope 254Cf, which decays with spontaneous fission even in 99% of
cases; however, its disadvantage is more difficult preparation
and shorter half-life T1/2 = 60.5 days.
Transurans decay
radioactively through entire decay series (similar to uranium and thorium - see Figure
1.4.1 above ),
where individual daughter products show alpha and beta
radioactivity and excited nuclei emit gamma radiation. The end
product is stable isotopes of lead or bismuth (if overlook a the very low radioactivity of 209Bi with a half-life
of 1.9.1019 years).
Below is a very brief
table of some of the most important radionuclides, which serves
only to illustrate the theoretical analysis of radioactivity in §1.2 and the properties of radionuclides in this §1.4. A
complete and detailed overview of radionuclides, with decay
schemes and all their characteristics, can be found, for example,
in the excellent "Table of isotopes"
by Lederer, Hollander, Perlman (new
electronic version http://ie.lbl.gov/toipdf/toi20
.pdf ).
Table of
the most frequently used radionuclides
| Radionuclide |
Half time T 1/2 |
Method of
disintegration |
Energy [keV]
Ea or Ebmax |
Energy [ keV ]
gamma E g |
The most common
method of production |
Use |
| 1
n 0 |
13 min |
b - |
782 |
- |
reactor 235
U,
( a , n) |
nuclear
analysis |
| 3 H 1 |
12.3 years |
b - |
18.6 |
- |
6 Li (n, a ) 3
H |
biology |
| 14
C 6 |
5730 years |
b - |
156 |
- |
14 N (n, p) 14
C |
biology
analysis |
| 18
F 9 |
110 min. |
b + (97%)
EC (3%) |
633 |
511 (194%)
(annihilation) |
18 O (p, n) 18
F |
nuclear
medicine |
| 32
P 15 |
14.3 days |
b - |
1710 |
- |
32 S (n, p) 32
P |
nuclear
medicine |
| 40
K 19 |
1.28. 10 9
years |
b - (89%) EC (11%)
|
1314 |
1460 (11%) |
natural
radionuclide |
isotope.
dating |
| 51
Cr 24 |
27.7 days |
EC |
- |
320 (9.8%) |
50 Cr (n, g ) 51
Cr |
nuclear
medicine |
| 57
Co 27 |
271 days |
EC |
- |
14 (9%)
122 (86%)
136 (11%)
692 (0.15%) |
56 Fe (d, n)
57 Co
56 Fe (p, g
) 57 Co
55 Mn ( a , 2n) 57 Co |
gamma source |
| 58
Co 27 |
70.8 days |
b + , EC |
- |
511 (30%)
810 (99%)
865 (1.5%)
1670 (0.6%) |
55 Mn ( a , n) 58
Co |
biology,
nuclear
medicine |
| 60
Co 27 |
5,271 years |
b - |
310 (99.88%)
1480 (0.12%) |
1173 (100%)
1332 (100%) |
59 Co (n, g ) 60
Co |
gamma source |
| 67
Ga 31 |
3.26 days |
EC |
- |
93 (40%)
184 (20%)
300 (17%)
393 (5%) |
68 Zn (p,
2n) 67 Ga |
nuclear
medicine |
| 68
Ga 31 |
68 min. |
b + (89%)
EC (11%) |
|
511 (178%)
(annihilation) |
68 Zn (p, n)
68 Ga |
nuclear
medicine |
81
Rb 37
êêêê |
4.6 hours
êêê |
EC is
|
- |
190 (66%)
446 (19%)
|
79
Br ( a , 2n) 81 Rb
|
generator
81m Kr
êêê |
| 81m
Kr 36 |
13 sec. |
IT |
- |
190 (67%) |
81
Rb ® 81m Kr |
lung scintigraphy |
| 90
Sr 38 |
28.8 years |
b - |
546 |
- |
235 U (n, f)
90 Sr |
|
| 90
Y 39 |
64 hours |
b - |
2280 |
- |
90 Sr ® 90
Y
89 Y (n, g
) 90 Y |
nuclear
medicine |
99
Mo 42
êêêê |
66 hours
êêê |
b -
êêê |
436 (17%)
1214 (83%) |
140 (6%)
181 (7%)
740 (13%)
778 (5%) |
98
Mo (n, g ) 99 Mo
235 U (n, f) 99 Mo
|
generator
99m Tc
êêê |
| 99m
Tc 43 |
6 o'clock |
IT |
- |
140 (90%) |
99
Mo ® 99m Tc |
scintigraphy |
| 111
In 49 |
2.8 days |
EC |
- |
171 (90%)
245 (94%) |
111 Cd (p,
n) 111 In |
nuclear
medicine |
| 123
I 53 |
13.2 hours |
EC |
- |
K a
27 (71%)
K b 31
(16%)
159 (83%) |
121 Sb ( a , 2n) 123
I |
nuclear
medicine |
| 125
I 53 |
60 days |
EC |
- |
K a
27 (112%)
K b 31
(25%)
35 (6.5%) |
124 Xe (n, g ) 125
Xe
â EC
125 I |
nuclear
medicine
(RIA) |
| 131
I 53 |
8.04 days |
b - |
334 (7.5%)
606 (90%) |
80 (2.5%)
284 (6%)
364 (81%)
637 (7%)
723 (2%) |
130 Te (n, g ) 131
Te
235 U (n, f) 131 Te
b - (25min) :
131 Te ® 131 J |
nuclear
medicine |
| 133
Xe 54 |
5.3 days |
b - |
346 |
K a
31 (39%)
K b 35
(9%)
81 (37%) |
235 U (n, f)
133 Xe |
nuclear
medicine |
| 137
Cs 53 |
30 years |
b - |
1176 |
K a 32 (6%) K b 36 (1.5%) g 662 (85%)
|
235 U (n, f)
137 Cs |
gamma source |
| |
|
|
|
|
|
|
| |
|
|
|
|
|
|
| |
|
|
|
|
|
|
| |
|
|
|
|
|
|
| |
|
|
|
|
|
|
| 192
Ir 77 |
74.2 days |
b - (95%)
EC (5%) |
240 (8%)
536 (41%)
672 (46%) |
296 (29%)
308 (30%)
317 (81%)
468 (49%)
604 (9%)
612 (6%) |
191 Ir (n, g ) 192
Ir
192 Os (d, 2
n) 192 Ir |
gamma source |
| 201
Tl 81 |
73 hours |
EC |
- |
K a
70 (74%)
K b 80
(21%)
135 (2.6%)
167 (9%) |
203 Tl (p,
3n) 201 Pb
â EC
201 Tl |
nuclear
medicine |
| |
|
|
|
|
|
|
| 226
Ra 88 |
1602
years |
a |
4782 |
186 (4%) |
natural
radionuclide |
alpha source |
| 232
Th 90 |
1.41. 10 10
years |
a |
4011 |
|
natural
radionuclide |
potential
nuclear
fuel |
| 235
U 92 |
7.1. 10 8
years |
a |
4580 |
143 (11%)
185 (54%)
204 (5%)
|
natural
radionuclide |
fissile
material |
| 238
U 92 |
4.51. 10 9
years |
a |
4195 |
|
natural
radionuclide |
nuclear
reactor |
| 239
Pu 94 |
2.44. 10 4
years |
a |
5160 |
|
238 U (n, g ) 239 U ® (b - ) 239 Np 3
® (b - ® 239 Pu
|
fissile
material |
| 241
Am 95 |
458 years |
a |
5486 |
L a 13.9 (14%)
L b 17.8
(20%)
L g 20.8 (
5%) g 26.4 ( 3%) g 59.6 (36%)
|
239 Pu (n, g ) 240
Pu
(n, g ) 241 Pu
â b -
241 Am |
alpha
and
gamma source |
| 252 Cf 98 |
2.65 years |
a (97%)
spont.
cleavage
(3%)
® n |
a:
6119 (97%)
+
cleavage fragm.
+
neutrons |
- |
238 U, 239
Pu
(n, g ), (n, g ), ....
..., (n, g ), .. b - ...
â
252 Cf |
neutron source |
Note: Pairs of parent ® daughter radionuclide used in
generatorsare marked with a colored
background.
Vojtech Ullmann